 ACME SMART PUBLICATION
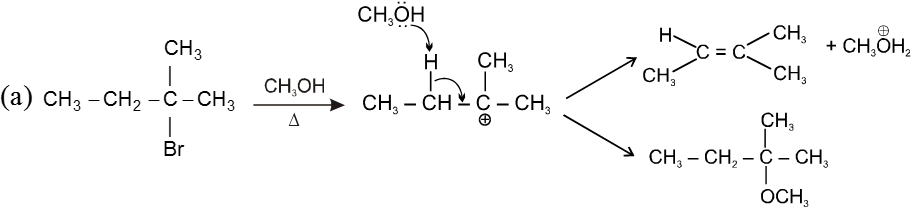
ACME SMART PUBLICATION
1. Coordination Entity/Coordination Sphere :
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 10:
coordination compounds
Addition Compounds :
They are formed by the combination of two or more stable compounds in stoichiometric ratio.
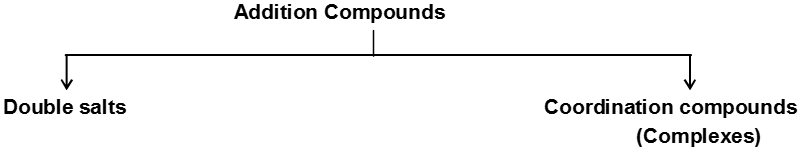
Double salts :
Those addition compounds which lose their identity in solution are called double salts.
K2SO4+ Al2(SO4)3 + 24H2O ® K2SO4 . Al2(SO4)3 . 24H2O![]() 2K+ (aq.) + 2Al+3 (aq.) + 4SO42– (aq.)
2K+ (aq.) + 2Al+3 (aq.) + 4SO42– (aq.)
Other examples are carnallite (KCl. MgCl2 . 6H2O), Mohr’s salt [FeSO4 . (NH4)2SO4 . 6H2O],
Potash alum [KAl(SO4)2.12H2O] etc.
Coordination Compounds :
Those addition compounds which retain their identity (i.e. doesn’t lose their identity) in solution are called coordination compounds.
Fe(CN)2 + 4KCN —® Fe(CN)2 . 4KCN or K4 [Fe(CN)6] (aq.) ![]() 4K+ (aq.) + [Fe(CN)6] 4-(aq.)
4K+ (aq.) + [Fe(CN)6] 4-(aq.)
Other examples are,
[Cu(NH3)4]SO4 (aq.) ![]() [Cu(NH3)4]2+ (aq.) + SO42– (aq.)
[Cu(NH3)4]2+ (aq.) + SO42– (aq.)
K2[Zn(CN)4] (aq.) ![]() 2K+ (aq.) + [Zn(CN)4]2– (aq.)
2K+ (aq.) + [Zn(CN)4]2– (aq.)
Coordination compound is defined as a species in which metal atom or ion is attached to group of neutral molecules / ions by coordinate covalent bonds.
Coordination Entity/Coordination Sphere :
A coordination entity constitutes a central atom/ion, usually of a metal, to which are attached a fixed number of other atoms or groups each of which is called a ligand. It may be neutral or charged. Examples being : [Co(NH3)6]3+, [PtCl4]2–, [Fe(CN)6]3–, [NiCl2(OH2)4] and (NH3), (Cl–), (CN–), (H2O) are the ligands.
The central atom/ion and the ligands attached to it are enclosed in square bracket and is collectively called as coordination sphere.
Note : The remaining ions apart from complex ions i.e. outside the coordination sphere are called counter ions, free ions or ionisable ions. For example, in K4[Fe(CN)6], the potassium (K+) ion is counter ion of coordination entity [Fe(CN)6]4–.
Central Atom/Ion :
In a coordination entity–the atom/ion to which are bound a fixed number of ligands in a definite geometrical arrangement around it, is called the central atom or ion. For example, the central atom/ion in the coordination entities : [NiCl2(OH2)4], [CoCl(NH3)5]2+ and [Fe(CN)6]3– are Ni2+, Co3+ and Fe3+, respectively. These central atoms / ions are also referred to as Lewis acids.
Ligands :
The neutral molecules, anions or cations which are directly linked with central metal atom or ion in the coordination entity are called ligands.
These may be simple ions such as Br–, small molecules such as H2O or NH3, larger molecules such as H2NCH2CH2NH2 or N(CH2CH2NH2)3 or even macromolecules such as proteins.
When a ligand is attached to a metal atom ion through a single donor atom, as with Cl–, H2O or NH3, the ligand is said to be unidentate. Similarly when a ligand is bound through two donor atoms, as in H2NCH2CH2NH2 (ethane-1, 2-diamine) or C2O42– (oxalate), the ligand is said to be bidentate and when several donor atoms are present in a single ligand as in N (CH2CH2NH2)3 or ethylenediaminetetraacetic acid (EDTA), the ligand is said to be polydentate.
Ambidentate Ligand :
Ligands which can ligate through two different atoms present in it are called ambidentate ligands. Examples of such ligands are the CN–, NO2– and SCN¯ ions. NO2– ion can coordinate through either the nitrogen or the oxygen atoms to a central metal atom/ion. Similarly, SCN¯ ion can coordinate through the sulphur or nitrogen atom. Such possibilities give rise to linkage isomerism in coordination compounds. For example,
![]() nitrito-N
nitrito-N
M ¬ O —N=O nitrito-O
M¬ SCN thiocyanato or thiocyanato-S
M¬ NCS isothiocyanato or thiocyanato-N
Chelate ligand :
Chelate ligand is a di or polydentate ligand which uses its two or more donor atoms to bind a single metal ion producing a ring. The complex formed is referred to as a chelate complex and the process of chelate formation is called chelation. The number of such ligating groups is called the denticity of the ligand.
1. Alkyl Halides
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 11: Reaction Mechanism
Introduction :
Alkyl halides :
There are three major classes of organohalogen compounds ; the alkyl halides, the vinyl halides, and the aryl halides.
An alkyl halide simply has a halogen atoms bonded to one of the sp3 hybrid carbon atoms of an alkyl group. (A vinyl halide or Aryl halide has a halogen atom bonded to one of the sp2 hybrid carbon atoms of an aromatic ring. They are different from alkyl halides because their bonding and hybridization are different.)
Classification of halides :
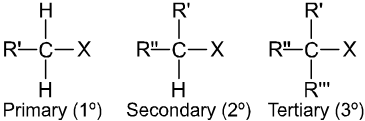
(a) Alkyl halides or haloalkanes (R—X) Compounds Containing sp3 C–X Bond :
They are classified as primary, secondary or tertiary according to the nature of carbon to which halogen is attached.
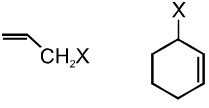
(b) Allylic halides :
These are the compounds in which the halogen atom is bonded to an sp3–hybridised carbon atom next to carbon-carbon double bond (C=C) i.e. to an allylic carbon.
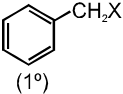
(c) Benzylic halides :
These are the compounds in which the halogen atom is bonded to an sp3–hybridised carbon atom next to an aromatic ring.
(d) Compounds Containing sp2 C–X Bond : Vinylic halides ![]() Aryl halides
Aryl halides ![]()
Structure of alkyl halide :
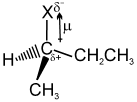
The carbon-halogen bond in an alkyl halide is polar because halogen atoms are more electronegative than carbon atoms. Most reactions of alkyl halides result from breaking this polarized bond. The carbon atom has a partial positive charge, making it some what electrophilic.
1. Strucutre and bonding in aldehydes and ketones
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 12: Aldehydes, Ketones and Carboxylic Acids
Aldehydes and Ketones
Introduction
Aldehydes & ketones have general formula CnH2nO and contains >C = O group. Thus aldehydes (R–CHO) and ketones (R–CO–R) are collectively called as carbonyl compounds. Aldehyde is always at terminal position while ketone is never at terminal position.
Structure and bonding in aldehydes and ketones
The carbonyl carbon atom is sp2 hybridized. The un hybridized p-orbital overlaps with a p-orbital of oxygen to form a pi bond. The double bond between carbon and oxygen is shorter, stronger, and polarized.
Orbital diagram for the formation of carbonyl group is as follows:
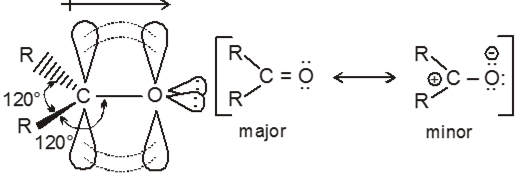
This polarity confirms that there is nucleophilic addition reaction takes place in carbonyl compound.
The double bond of the carbonyl group has a large dipole moment because oxygen is more electronegative than carbon.
Carbonyl carbon act as an electrophile (Lewis acid)
Carbonyl oxygen act as a nucleophile (Lewis base)
1. Amines
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 13: Organic Compounds contaning Nitrogen
Amines
Amines are derivatives of ammonia in which one or more hydrogen atoms are replaced by alkyl group(s).
Amines are classified as primary, secondary and tertiary depending on the number of alkyl groups attached to nitrogen atom.

General methods of preparation
(1) Ammonolysis of alkyl halides and alcohols :
(a) From Ammonolysis of alkyl halides (Hofmann's ammonolysis) : When an aqueous solution of ammonia is heated with alkyl halide all the three types of amines and quaternary ammonium salt are formed.
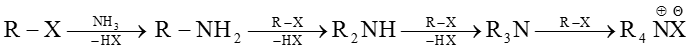
(Quaternary ammonium salt)
If ammonia is taken in excess, 1° amine is the main product.
(b) Ammonolysis of alcohols : When ROH and NH3 are passed over At2O3 or ThO2 at 350°
Call the three types of amines are formed.
![]()
- Quaternary ammonium hydroxide is not formed.
- If excess of ammonia is used, then main product will be primary amine.
(2) By reduction :
(a) With RCONH2 : RCONH2 ![]() RCH2NH2
RCH2NH2
(b) With RCN : RCN + 4[H] ![]() RCH2NH2
RCH2NH2
This reaction (b) is called mendius reaction.
The reduction of alkyl isocynides gives secondary amines.
R–NC + 4[H]![]() RNHCH3
RNHCH3
(c) With Oximes : R–CH=N–OH + 4[H]![]() RCH2–NH2 + H2O
RCH2–NH2 + H2O
(d) With RNO2 : RNO2 + 6[H] ![]() RNH2 + 2H2O
RNH2 + 2H2O
Sn/HCl is used in laboratory preparation
(3) By hydrolysis of :
(a) R–NC : Alkyl isocyanide undergoes hydrolysis with mineral acid and forms alkyl amine.
R–NC + 2H2O![]() RNH2 + HCOOH
RNH2 + HCOOH
(b) RNCO : Alkyl isocyarlate undergoes hydrolysis on heating with KOH and form alkyl amine.
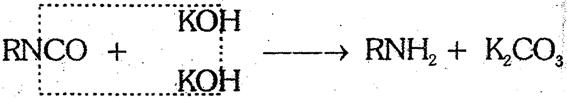
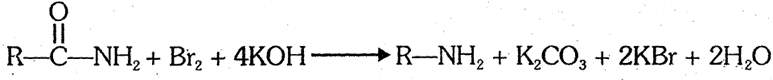
(4) By Hofmann's bromamide reaction (Hofmann's Hypobromite reaction) : This is a general method for the conversion of alkanamides into primary amines having one less carbon.
(5) From Grignard reagent : Alkyl magnesium iodide reacts with chloramine to yield alkyl amine.
![]()
(6) Gabriel phthalimide synthesis : Phthalimide is first treated with KOH to obtain potassium phthalimide which is then treated with alkyl iodide. Then alkyl phthalimide on hydrolysis yields alkylamine. This method is used in the formation of pure aliphatic primary amines.
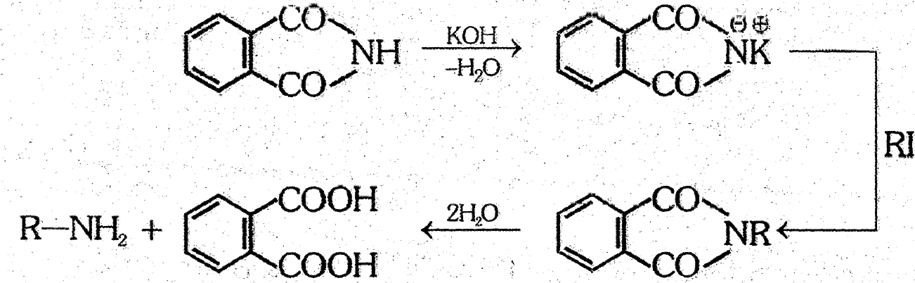
Phthalic acid
- Aniline is not formed by this reaction.
(7) Curtius reaction :

(8) Schmidt reaction : In presence of conc. H2SO4 alkanoic acid reacts with hydrazoic acid (N3H) followed by hydrolysis to yield alkylamine.
![]()
1. Carbohydrates
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 14: biomolecules & polymers
Carbohydrates (Saccharides) : (Latin word : saccharum = sugar)
Concept
Generally, carbohydrates are substances with the general formula Cx(H2O)y , and were therefore called carboydrates (hydrates of carbon) because they contained hydrogen and oxygen in the same proportion as in water. How ever, a number of compounds have been discovered which are carbohydrates by chemical behaviour but do not conform to the formula Cx(H2O)y e.g ,2-deoxyribose, C5H10O4.
Hence, carbohydrates are biopolymers of polyhydroxy aldehyde or polyhydroxy ketones. “There monomeric polyhydroxy aldehydes or ketones can also exist in hemiocetal and acetal forms in cyclic structures.”
# Note:
Almost all of these compounds are chiral & optically active. “An exception of this is 1,3-dihydroxypropanone, CH2OHCOCH2OH”.
2 Classification of carbohydrates :
(A) Classification on the basis of number of hydrolysed products

(B) On the Basic of functional group
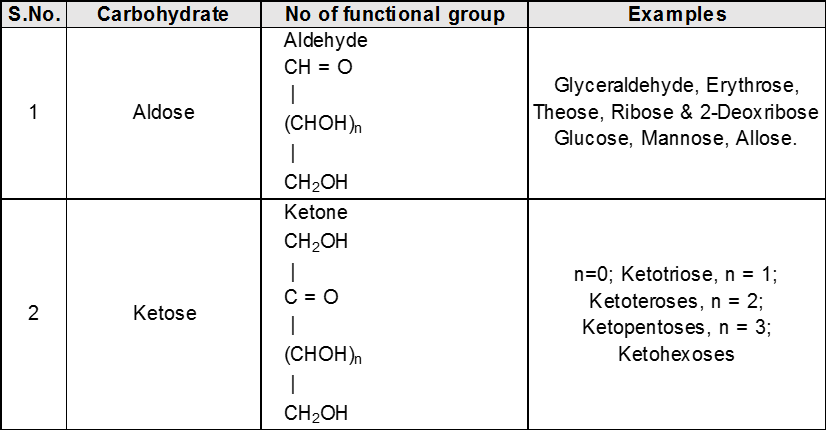
(C) On basis of carbon atoms.

Configuration of the D-Aldoses
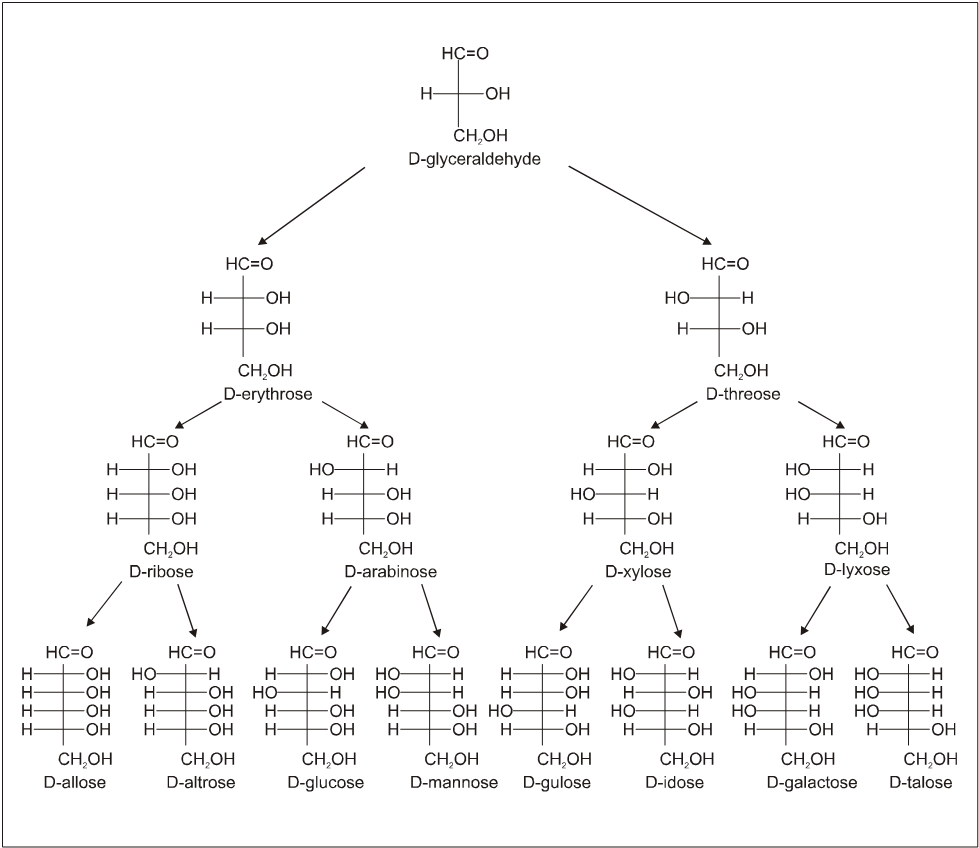
3. Structure :
(i) All form of Aldose or ketose may exist in open chain form as well as in cyclic pyramose/furanose form.
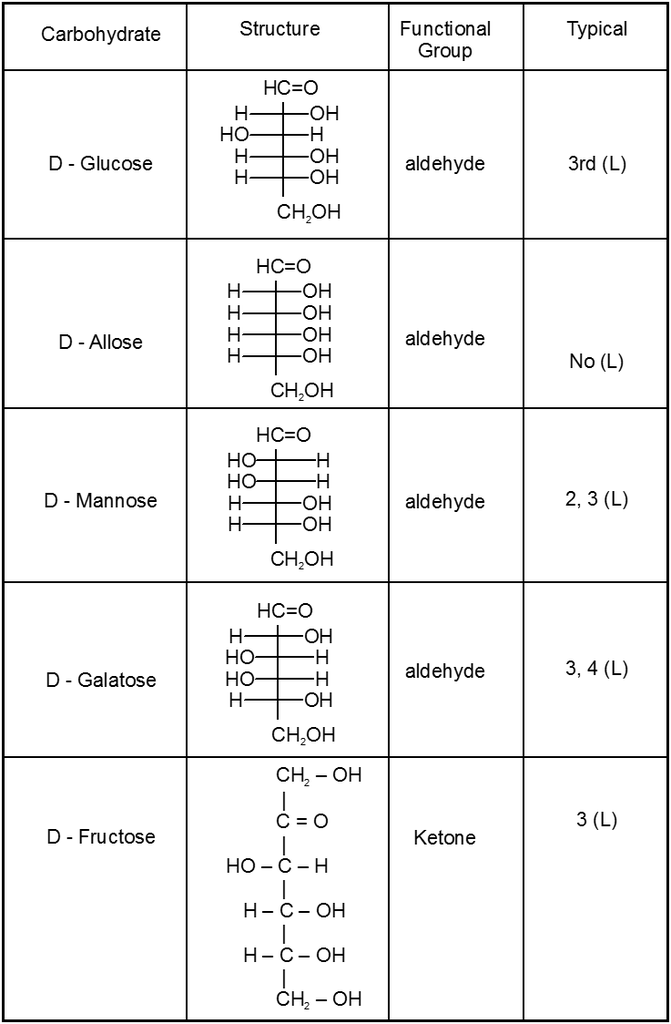
Cyclie structure of monosaccharide

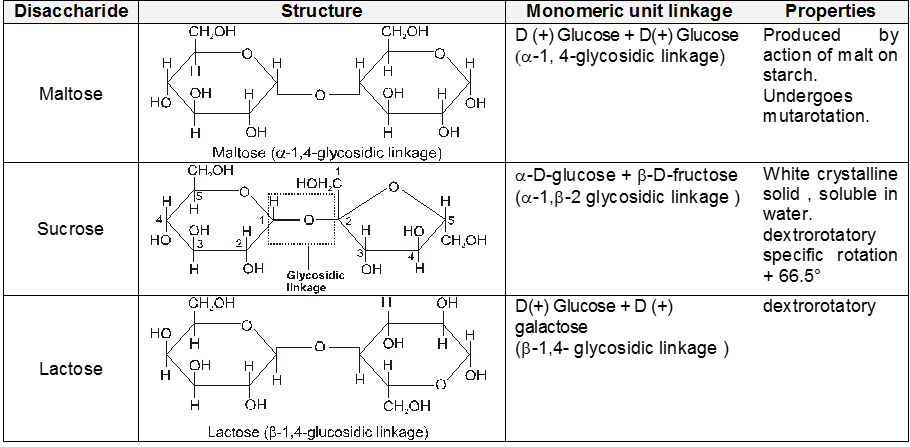
Disaccharide
Polysaccharide
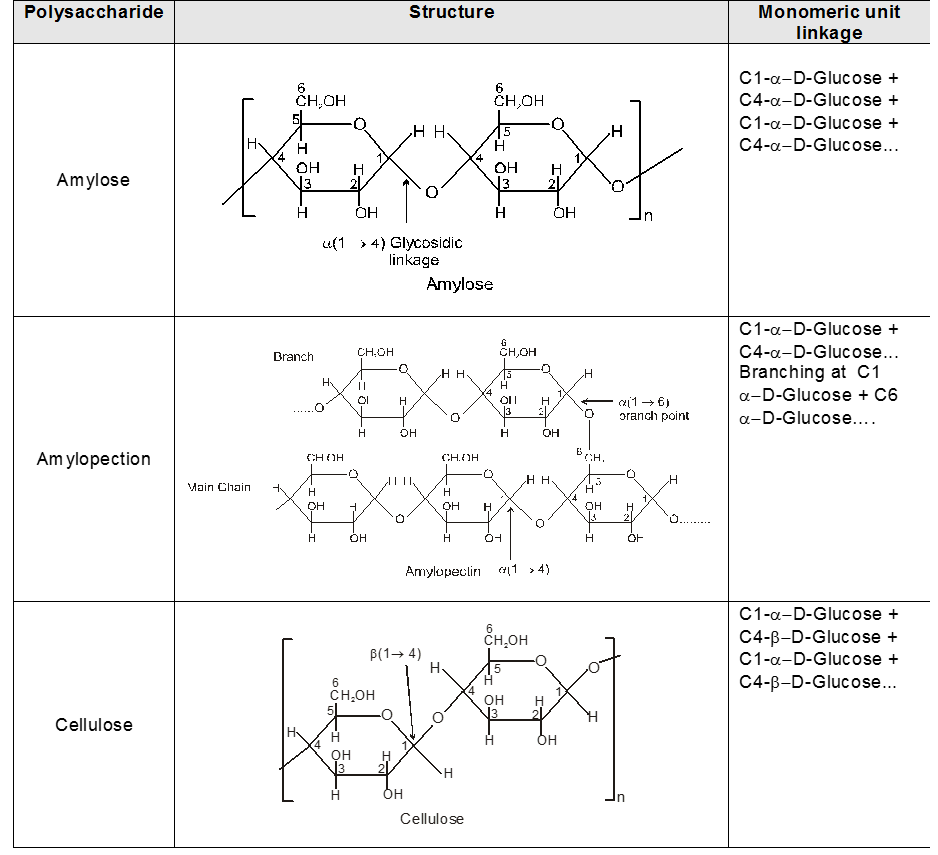
INTER CONVERSION OF a and b
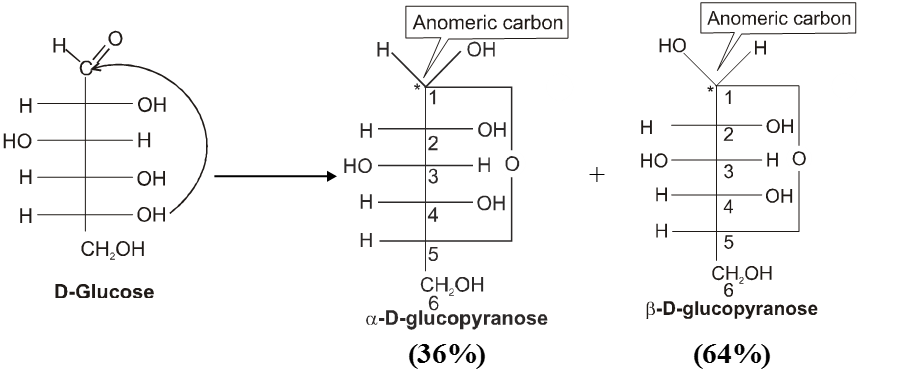
Haworth projection
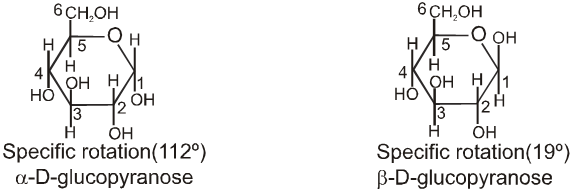
Chair conformation structures :
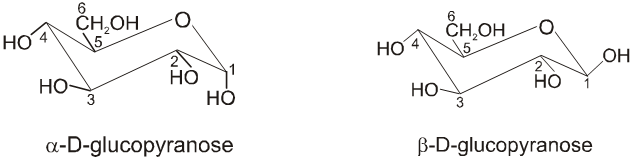
Anomers :
Anomers are diastereomers that differ in the configuration at the acetal or hemiacetal C atom of a sugar in its cyclic form or Anomers are epimers whose conformations differ only about C-1.
For example, a D(+) and b -D(+) glucose are anomers. a-D(–) and b-D(–) fructose are anomers.
Epimers :
Diastereomers with more than one stereocentre that differ in the configuration about only one stereocentre are called epimers.
i. D-Glyceraldehyde and L-glyceraldehyde (this pair is an enantiomer as well as an epimer).
ii. D-Erythrose and L-threose are epimers.
iii. D-glucose and D-galactose are C-4 epimers and
iv. D-idose and D-talose are C-3 epimers.
v. D-glucose and D-mannose are C-2 epimers.
vi. Epimerisation of glucose at C-2 gives mannose.
vii. Epimerisation of glucose at C-3 gives allose.
viii. Epimerisation of glucose at C-4 gives galactose.
Action of alkalies :
(a) With conc. NaOH glucose first turns yellow resinify. Fructose is not effected by conc. NaOH
(b) With dil. NaOH/KOH both Glucose & Fructose undergo a rearrangement to give equilibrium mixture of three sugars.
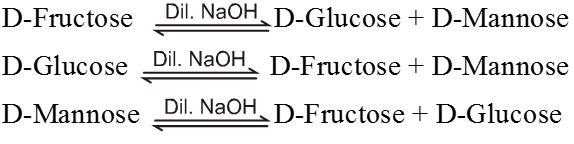
This rearrangement is known as Lobry De-bryn. van Ekenstein rearrangement.
4. Properties of Anomers : Mutarotation
When one of the pure glucose anomers dissolve in water, an intersting change in the specific rotation is observed. When the a-anomer dissolves, its specific rotation gradually decreases from an initial value of +112º to +52.7º. When the pure b- anomer dissolves, its specific rotation gradually increases from +19º to the same value of +52.7º. This change (mutation) in the specific rotation is called mutarotation. What is happening is that each solution, initially containing only one anomeric form, undergoes equilibrium to the same mixture of a-and b-forms. The open chain forms is in intermediate in the process.
For mutarotation atleast one hemiacetal group must be present in the sugar therefore all reducing sugars will mutarotate.
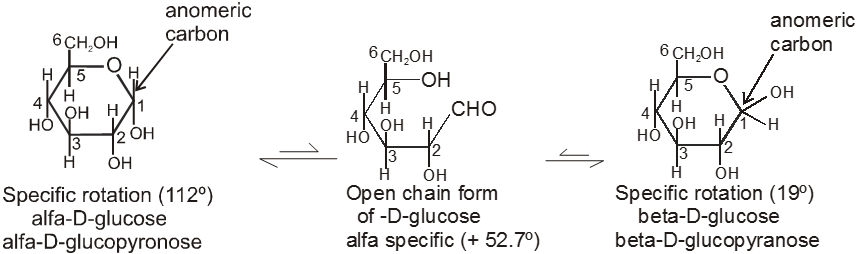
(C) Classification on the basis of Reducing and non Reducing properties :
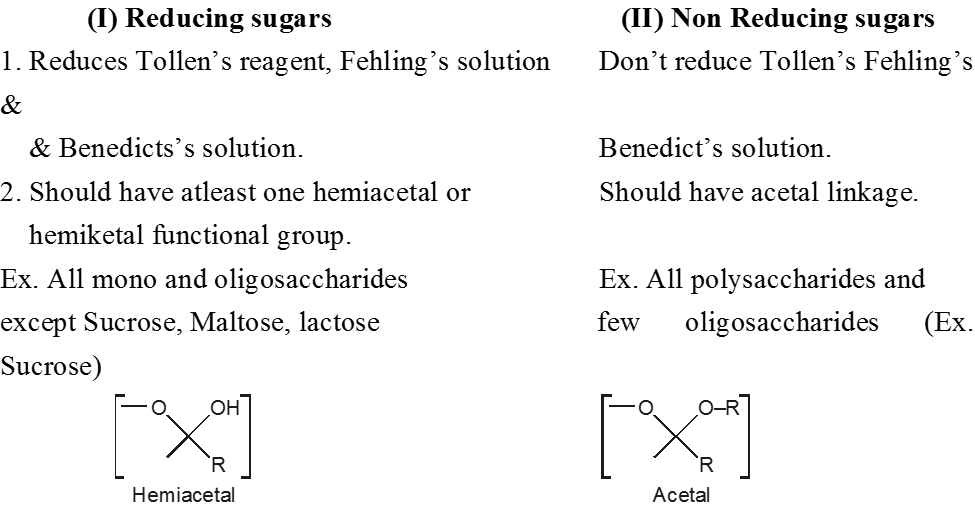
Monosaccharides :
(A) Glucose (C6H12O6) :
Glucose is the most common monosaccharide. It is known as Dextrose because it occurs in nature principally as the optically active dextrorotatory isomers. It is act as a reducing agent (reduces both Fehling’s solution and ammonical silver nitrate solution). When heated with sodium hydroxide, an aqueous solution of glucose turns brown. It is known as dextrose and found as grapes, honey, cane sugar, starch and cellulose.
Preparation :
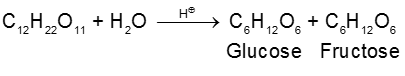
(1) By acid hydrolysis of cane sugar (a disaccharide) :
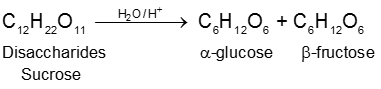
a-Glucose being much less soluble in alcohol than fructose separate out by crystallization on cooling.
(2) By enzymatic action over starch :
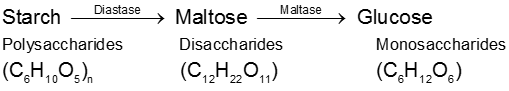
Physical properties :
(i) It is white crystalline solids having melting point 146ºC. It is readily soluble in water.
(ii) Glucose is sweet in taste and also optically active (dextro rotatory).
(iii) Glucose shows mutarotation.
Chemical properties :
(1) Reactions due to OH group :
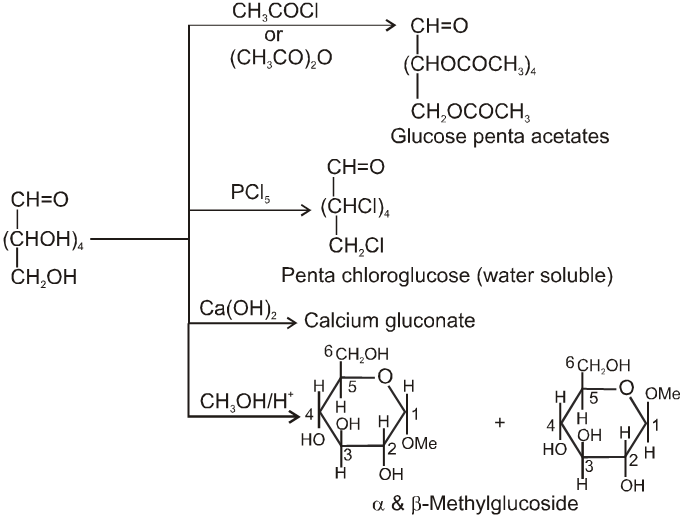
(2) Reactions due to –CHO group :

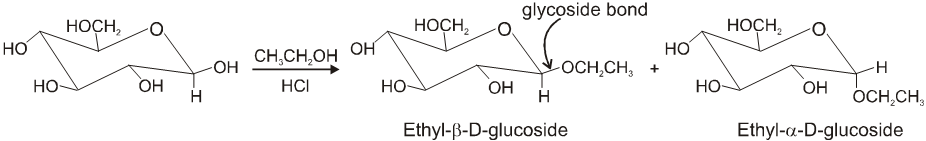
Glycosides-formation :
(B) Fructose :
- Also said to be fruit sugar
- It occur both in combined as well as free state.
- Fructose is named as fruit sugar because it is present in honey and most sweet fruit in free state.
- It is sweetest monosaccharide and present in cane sugar and insulin in combined state.
- It is also known as a-Laevulose i.e. natural occurring compound is laevorotatory
Preparation :
By acid hydrolysis of cane sugar :
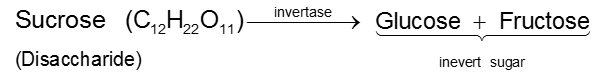
By enzymatic action of sucrose
Note: Glucose and fructose obtained by acid hydrolysis of sucrose can be separated by treating with Ca(OH)2 which forms calcium glucosate & calcium fructosate. Calcium fructosate being water insoluble. It is seperated out easily
Physical properties :
(1) It is colourless crystalline solid.
(2) It is soluble in water but insoluble in ether ketone and benzene.
(3) It is pentahydroxy ketone and shows mutarotation like glucose.
(4) It is reducing sugar.
Chemical Properties :
Due to OH group:
(1) It froms fructose penta acetate with acetyl chloride :
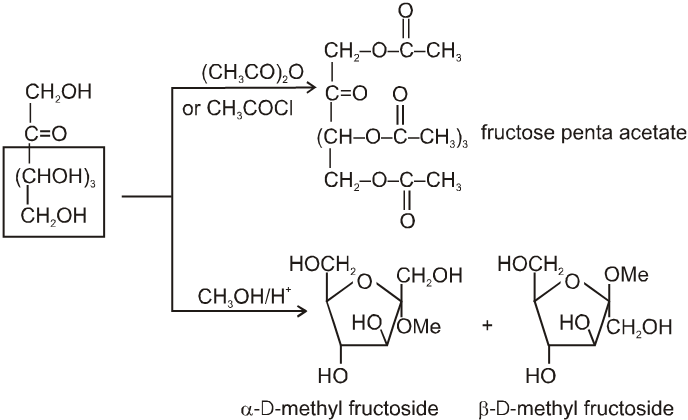
(2) Reaction due to keto group :
It also form osazone with excess of phenyl hydrazine thus we can say that osazone formation is characteristic of a-hydroxy carbonyl compounds.
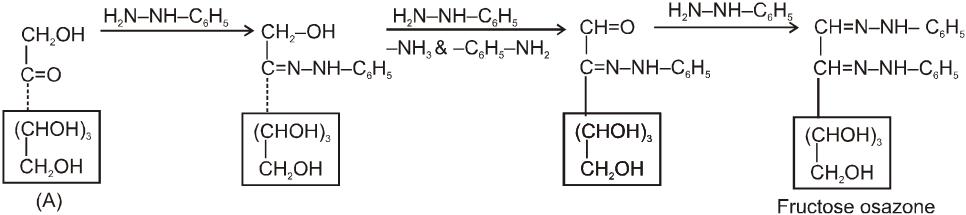
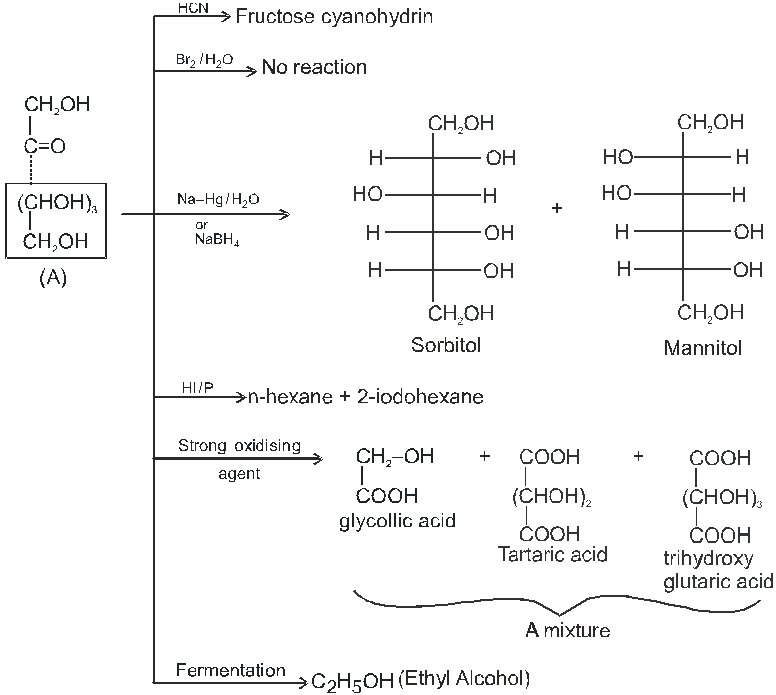
Note : 1. Unlike other ketones, fructose can reduced Fehling sol. and Tollen’s reagents it is probably due to formation of an equilibrium between glucose, mannose and fructose.
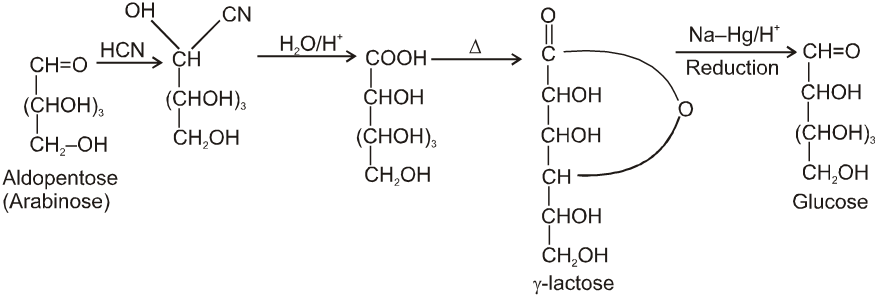
Chain lengthing :
Chain shortening :
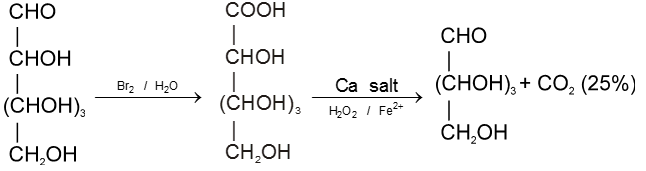
Disaccharides :-
(A) Sucrose : (Sucrose, Invert-sugar C12H22O11)
(a) Sucrose (Cane sugar) ![]() a–glucose + b-fructose
a–glucose + b-fructose
Formation of sucrose (C12H22O11)
a–D-glucose + b-D-fructose
C1 C2
Pyranose form Furanose form
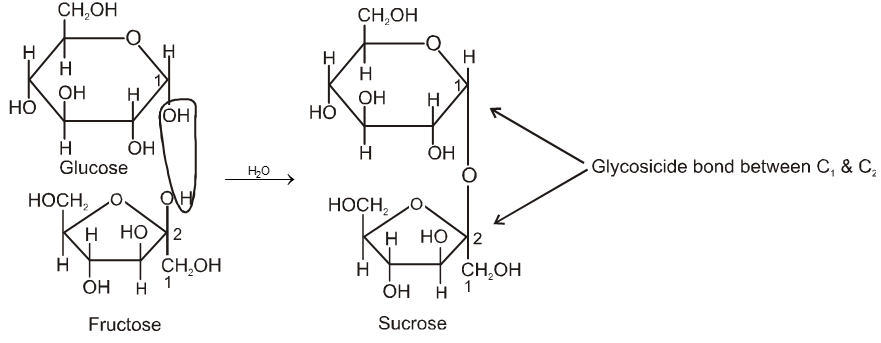
Two monosaccharides are joined together by an oxide linkage formed by loss of water molecule. Such linkage through oxygen atom is called glycosidic linkage.
In sucrose linkage in between C1 of a-glucose and C2 of b-fructose. Since the reducing group of glucose & fructose are involved in glycosidic bond formation, sucrose is non reducing sugar.
(i) On hydrolysis with dilute acids sucrose yields an equimolecular mixture of D(+)-glucose and
D(–)-fructose :
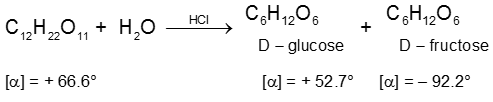
Since D(–)-fructose has a greater specific rotation than D(+)-glucose, the resulting mixture is laevorotatory. Because of this, hydrolysis of cane-sugar is known as the inversion of cane-sugar or Inversion of sucrose (this is not to be confused with the Walden inversion), and the mixture of sugars are known as invert sugar Ex. D - Glucose & D-Fructose. The inversion (i.e., hydrolysis) of cane-sugar may also be effected by the enzyme invertase which is found in yeast.
(ii) Sucrose is not a reducing sugar, e.g., it will not reduce Fehling’s solution or Tollen’s reagnet. It does not form an oxime or an osazone, and does not undergo mutarotation. This indicates that hemiacetal group is not present in the rings.
Polysaccharides :-
(A) Starch (C6H10O5)n :
(i) Starch is the main contributor of carbohydrates in our diet. It exists exclusively in plants, stored in the seeds, roots, and fibres as food reserve. Example rice, potato.
(ii) Both amylose and amylopectin are composed of D-glucose units.
(iii) The amylose molecule is made up of D-glucose unit joined by a-glycosidic linkages between C-1 of one glucose unit and C-4 of the next glucose unit. The number of D-glucose units in amylose range from 60-300.
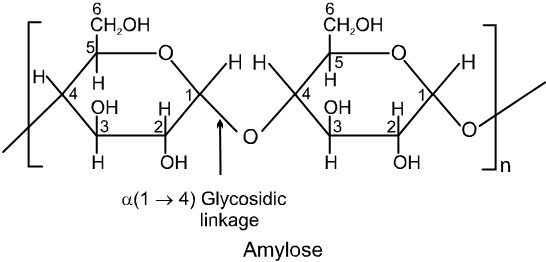
(iv) Amylopectin has a branched-chain structure. It is composed of chains of 25 to 30 D-glucose units joined by a-glycosidic linkages between C-1 to one glucose unit and C-4 of the next glucose unit. These chains are in turn connected to each other by 1, 6-linkages.
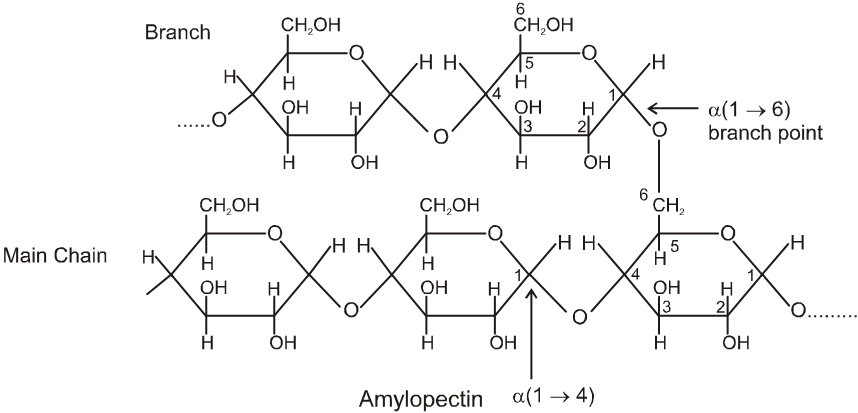
The solution of a-amylose gives a blue colour with iodine.
The solution of Amylopectin gives a violet colour with iodine :
(B) Cellulose, (C6H10O5)n :
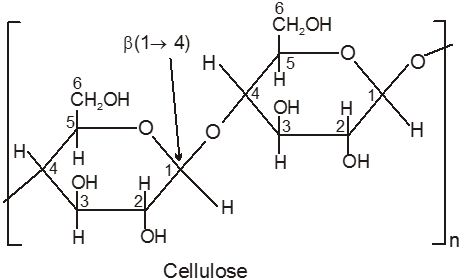
(i) Cellulose is the main structural material of trees and other plants. Wood is 50% cellulose, while cotton and wool are almost pure cellulose. Other sources of cellulose are straw, corncobs, bagasse, and similar agricultural wastes.
(ii) Artificial silk, rayon, is used collectively to cover all synthetic or manufactured fibres from cellulose.
(iii) The nitrates are prepared by the reaction of cellulose with a mixture of nitric and sulphuric acids, and the degree of ‘nitration’ depends on the concentrations of the acids and the time of the reaction. Cellulose trinitrate (12.2 – 13.2%N) is known as gun-cotton and is used in the manufacture of blasting explosives and smokeless powders.
(iv) Non-reducing sugar.
1. Drugs and their Classification
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 15: Chemistry in Everyday Life
1. Introduction to Drugs and Chemotherapy :
In general the drug may be defined as the substances used in the prevention, diagnosis, treatment or cure of disease in man or animals.
When administered to the ailing individual, its action should be localised at the site where it is desired to act. (In actual practice, there is no drug which behaves in this manner).
Chemotherapy :
“The use of chemicals to destroy infectious micro organisms without causing any injury to the host is called as chemotherapy” .
Drugs and Classification of drugs
(a) On the basis of Pharmacological effect
(b) On the basis of drug action
(c) On the basis of chemical structure
(d) On the basis of molecular targets
Drug Target Interaction :
Macromolecules of biological origin perform various functions in the body for example proteins which perform the role of biological catalysts in the body are called enzymes, and those which are crucial to communication system in the body are called receptors.
Enzymes as Drug Targets :
(a) Catalystic action of enzymes :
In catalytic activity, enzymes perform two major functions as follows
(i) To hold the substrate for chemical reaction :
(ii) The second function of the enzyme is to provide functional group which will attacks the substrate to carry out chemical reaction.
Receptors as Drug Targets :
Receptors are proteins that are crucial to body’s communication process.
2. Denticity and Chelation
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Denticity and Chelation :
Table : 1
Common Monodentate Ligands
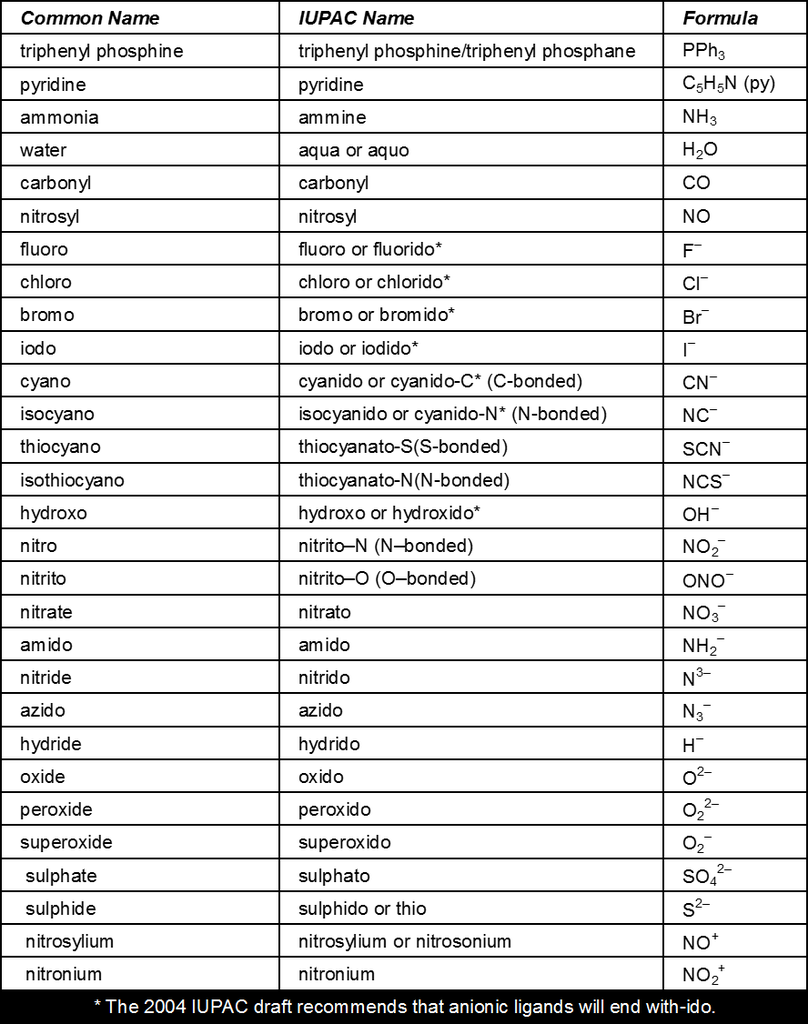
Table : 2
Common Chelating Amines

Coordination Number :
The coordination number of the central atom/ion is determined by the number of sigma bonds between the ligands and the central atom/ions i.e. the number of ligand donor atoms to which the metal is directly attached. Pi-bonds, if any, between the ligating atom and the central atom/ion are not considered for the determination of coordination number. The sigma bonding electrons may be indicated by a pair of dots, preceding the donor atom in the ligand formula as in :
[Co(NH3)6]3+, [Fe(CN)6]3–, [Ni(CO)4], [Co(Cl4)]2–.
Some common co-ordination number of important metals are as given below.

Coordination Polyhedron :
The spatial arrangement of the ligand atoms which are directly attached to the central atom/ion defines a coordination polyhedron about the central atom. Figure below shows the shapes of tetrahedral, square planar, octahedral, square pyramidal and trigonal bipyramidal coordination polyhedra. [Co(NH3)6]3+ has an octahedral geometry, while [PtCl4]2– and Ni(CO)4, are square planar and tetrahedral, respectively.
Oxidation number of Central Atom :
The oxidation number of the central atom is defined as the charge it would carry if all the ligands are removed along with the electron pairs that are shared with the central atom. Metal oxidation number is represented by a Roman numeral in parentheses following the name of the coordination entity. For example oxidation number of iron in [Fe(CN)6]3– is +3 and it is written as Fe(III).
Homoleptic and heteroleptic complexes
Complexes in which a metal is bound to only one type of donor groups, e.g., [Cr(NH3)6]3+, are known as homoleptic. Complexes in which a metal is bound to more than one type of donor groups. e.g., [Co(NH3)4Br2]+, are known as heteroleptic.
3. Nomenclature of Coordination Compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Nomenclature of Coordination Compounds
Writing the formulas of Mononuclear Coordination Entities :
The following rules are followed while writing the formulas :
(i) The central atom is placed first.
(ii) The ligands are then placed in alphabetical order. The placement of a ligand in the list does not depend on its charge.
(iii) Polydentate ligands are also placed alphabetically. In case of abbreviated ligand, the first letter of the abbreviation is used to determine the position of the ligand in the alphabetical order.
(iv) The formula for the entire coordination entity, whether charged or not, is enclosed in square brackets. When ligands are polyatomic, their formulas are enclosed in parentheses. Ligands abbreviations are also enclosed in parentheses.
(v) There should be no space between the ligands and the metal within a coordination sphere.
(vi) When the formula of a charged coordination entity is to be written without that of the counter ion, the charge is indicated outside the square brackets as a right superscript with the number before the sign. For example, [Co(H2O)6]3+, [Fe(CN)6]3– etc.
(vii) The charge of the cation(s) is balanced by the charge of the anion(s).
Writing the name of Mononuclear Coordination Compounds :
The following rules are followed when naming coordination compounds :
(i) Like simple salts the cation is named first in both positively and negatively charged coordination entities.
Examples :
[Ag(NH3)2]Cl,diamminesilver(I)chloride.K3[Fe(CN)6],potassium hexacyanidoferrate(III).
(ii) The ligands are named in an alphabetical order (according to the name of ligand, not the prefix) before the name of the central atom/ion.
Examples :
[Pt(NH3)BrCl(CH3NH2)], amminebromidochloridomethylamineplatinum(II).
[Co(H2O)2(ox)2]–, diaquabis(oxalato)cobaltate(III).
(iii) Names of the anionic ligands end in –o and those of neutral ligands are the same except aqua for H2O, ammine for NH3, carbonyl for CO, thiocarbonyl for CS and nitrosyl for NO. But names of cationic ligands end in–ium. The neutral an cationic are placed within enclosing marks ( ) .
Some more important examples of neutral and cationic ligands are :
tetraphosphorus — P4
dioxygen — O2
octasulphur — S8
(iv) Prefixes mono, di, tri, etc., are used to indicate the number of the one kind of ligands in the coordination entity. When the names of the ligands include a numerical prefix are complicated or whenever the use of normal prefixes creates some confusion, it is set off in parentheses and the second set of prefixes is used.

Example ;
[CoCl2(NH2CH2CH2NH2)2]+, dichloridobis(ethane-1,2-diamine)cobalt(III).
[NiCl2(PPh3)2], dichloridobis(triphenylphosphine)nickel(II).
(v) Oxidation state of the metal in cation, anion or neutral coordination entity is indicated by Roman numeral in the parentheses after the name of metal.
(vi) If the complex ion is a cation, the metal is named same as the element. For example, Co in a complex cation is called cobalt and Pt is called platinum. If the complex ion is an anion, the name of the metal ends with the suffix - ate. For example, Co in a complex anion, [Co(SCN)4]2– is called cobaltate. For some metals, the Latin names are used in the complex anions.

Examples ;
[Co(NH3)4Cl2]+, pentaamminechloridocobalt(III).
(NH4)2 [Co(SCN)4], ammonium tetrathiocyanato-S-cobaltate(II).
(vii) The neutral complex molecule is named similar to that of the complex cation.
Example ;
[CrCl3(py)3], trichloridotris(pyridine)chromium(III).
(viii) The prefixes cis- and trans- designate adjacent and opposite geometric locations. For examples,
[Pt(NH3)2Cl2], cis- and trans-diamminedichloridoplatinum(II),
[CoCl2(NH3)4]+, cis- and trans-tetraamminedichloridocobalt(III).
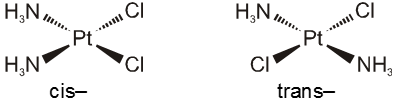
Effective Atomic Number Rule given by Sidgwick :
Effective Atomic Number (EAN) = No. of electron present on the metal atom/ion + No. of electrons donated by ligands to it.
OR
Effective Atomic Number (EAN) = Atomic no. of central metal – Oxidation state of central metal + No. of electrons donated by ligands.
The complexes in which the EAN of the central atom equals the atomic number of the next noble gas, are found to be extra stable.
Note : The EAN rule is generally found to be not valid in case of most of the complexes but in case of metal carbonyls this rule is found to be valid in all cases except one or two exceptions.
4. Werner's Theory
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Werner's Theory :
Werner in 1983 presented a theory known as Werner's coordination theory. More important postulates of this theory are :
Most element exhibit two types of valencies :
(a) Primary valency and
(b) Secondary valency.
(a) Primary valency :
This corresponds to oxidation state of the metal ion. This is also called principal, ionisable or ionic valency. It is satisfied by negative ions and its attachment with the central metal ion is shown by dotted lines.
(b) Secondary or auxiliary valency :
It is also termed as coordination number (usually abbreviated as CN) of the central metal ion. It is non-ionic or non-ionisable (i.e. coordinate covalent bond type). This is satisfied by either negative ions or neutral molecules having lone pair of electrons (e.g., H2O, NH3 etc.) or even sometimes by some positive groups. The ligands which satisfy the coordination number are directly attached to the metal atom or ion and shown by thick lines.
Every element tends to satisfy both its primary and secondary valencies. In order to meet this requirement a negative ion may often show a dual behaviour, i.e. it may satisfy both primary and secondary valencies since in every case the fulfillment of coordination number of the central metal ion appears essential. This dual behaviour is represented by both thick and dotted lines. For example, [CoCl(H2O)5]Cl2 is represented as
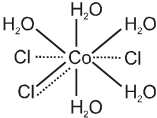
The ions/groups bound by the secondary valencies have characteristic spatial arrangements corresponding to different coordination number. In the modern terminology, such spatial arrangements are called coordination polyhedra and various possibilities are
C.N. = 2 linear
C.N. = 3 Triangular
C.N. = 4 tetrahedral or square planar
C.N. = 6 octahedral.
To distinguish between the two types of valencies, Werner introduced the square brackets [ ] to enclose those atoms making up the coordination complex and which are, therefore, not ionized.
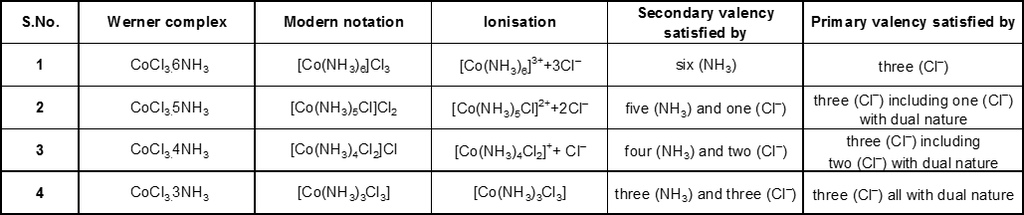
On the basis of the above postulates Werner formulated the coordination compounds, CoCl3 . 6NH3,
CoCl3 . 5NH3 and CoCl3 . 4NH3 as : [Co(NH3)6]Cl3, [Co(NH3)5Cl]Cl2 and [Co(NH3)4Cl2]Cl respectively; the species within the square brackets being the coordination entitles (complexes) and the ions outside the square brackets the counter ions. He further postulated that octahedral, square, planar and tetrahedral geometrical shapes are more common in coordination compounds of transition metals. Thus, [Co(NH3)6]3+, [CoCl(NH3)5]2+, [CoCl2(NH3)4]+ are octahedral entities, while [Ni(CO)4] and [PtCl4]2– are tetrahedral and square-planar, respectively.
5. Bonding in coordination compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Bonding in coordination compounds
Valence bond theory :
The valence bond theory, VBT, was extended to coordination compounds by Linus Pauling in 1931. The formation of a complex involves reaction between a lewis base (ligand) and a lewis acid (metal or metal ion) with the formation of a coordinate-covalent (or dative) bonds between them. The model utilizes hybridisation of (n-1) d, ns, np or ns, np, nd orbitals of metal atom or ion to yield a set of equivalent orbitals of definite geometry to account for the observed structures such as octahedral, square planar and tetrahedral, and magnetic properties of complexes. The number of unpaired electrons, measured by the magnetic moment of the compounds determines which d-orbitals are used.
These hybrid orbitals are allowed to overlap with ligand orbitals that can donate electron pairs for bonding.
Following table provides the types of hybridisation with different coordination number.

It is to be noted that the type of hybridisation of metal and shape of complex involved can be predicted conveniently, if some characteristic of the complex like magnetic nature, geometry or whether exhibits isomerism or not, etc., be known.
Coordination Number Six.
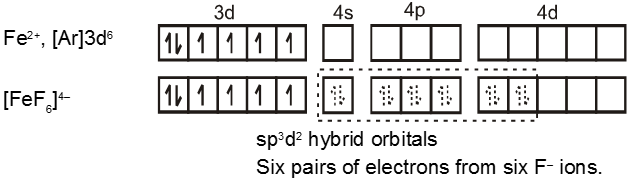
In the diamagnetic octahedral complex, [Co(NH3)6]3+, the cobalt ion is in +3 oxidation state and has the electronic configuration represented as shown below.
Co3+,[Ar]3d6 ![]()
[Co(NH3)6]3+ ![]()
![]()
Thus, the complex has octahedral geometry and is diamagnetic because of the absence of unpaired electron. Since in the formation of complex the inner d-orbital (3d) is used in hybridisation, the complex is called an inner orbital or low spin or spin paired complex.
The complex [FeF6]4– is paramagnetic and uses outer orbital (4d) in hybridisation (sp3d2) ; it is thus called as outer orbital or high spin or spin free complex. So :
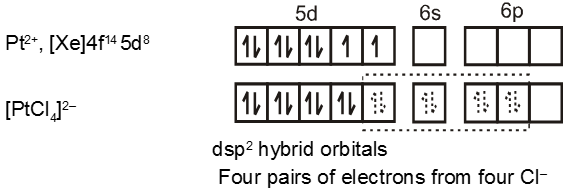
Coordination Number Four :
In the paramagnetic and tetrahedral complex [NiCl4]2–, the nickel is in +2 oxidation state and the ion has the electronic configuration 3d8. The hybridisation scheme is as shown in figure.
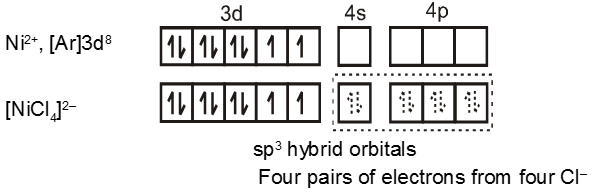
The compound is paramagnetic since it contains two unpaired electrons.
Similarly complex [Ni(CO)4] has tetrahedral geometry and is diamagnetic as it contains no unpaired electrons. The hybridisation scheme is as shown in figure.
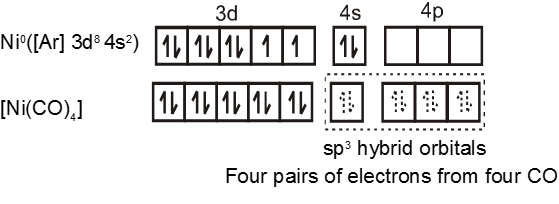
Complexes of Pd(II) and Pt (II) are usually four-coordinate, square planar, and diamagnetic
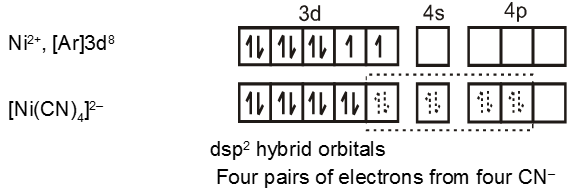
Similarly the hybridisation scheme for [Ni(CN)4]2– is as shown in figure.
While the valence bond theory, to a large extent, explains the formation, structures and magnetic behaviour of coordination compounds, it suffers from the following shortcomings :
1. A number of assumptions are involved.
2. There is no quantitative interpretation of magnetic data.
3. It has nothing to say about the spectral (colour) properties of coordination compounds.
4. It does not give a quantitative interpretation of the thermodynamic or kinetic stabilities of coordination compounds.
5. It does not make exact predictions regarding the tetrahedral and square-planar structures of 4-coordinate complexes.
6. It does not distinguish between strong and weak ligands.
6. Magnetic Properties of Coordination Compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Magnetic Properties of Coordination Compounds :
Additional information for understanding the nature of coordination entities is provided by magnetic
susceptibility measurements. We have noted that coordination compounds generally have partially filled d orbitals and as such they are expected to show characteristic magnetic properties depending upon the oxidation state, electron configuration, coordination number of the central metal and the nature of the ligand field. It is experimentally possible to determine the magnetic moments of coordination compounds which can be utilized for understanding the structures of these compounds.
The number of unpaired electrons in any complex can be easily calculated from the configuration of the metal ion, its coordination number and the nature of the ligands involved( strong or weak from the spectrochemical series) and after that the magnetic moment of the complexes can be easily calculated using
Magnetic Moment = ![]() Bohr Magneton
Bohr Magneton
For metal ions with upto three electrons in the d-orbitals like Ti3+, (d1) ; V3+ (d2) ; Cr3+ (d3) ; two vacant d-orbitals are easily available for octahedral hybridisation. The magnetic behaviour of these free ions and their coordination entities is similar. When more than three 3d electrons are present, like in Cr2+ and Mn3+ (d4) ; Mn2+ and Fe3+ (d5) ; Fe2+ and Co3+ (d6) ; the required two vacant orbitals for hybridisation is not directly available (as a consequence of Hund’s rules). Thus, for d4, d5 and d6 cases, two vacant d-orbitals are only available for hybridisation as a result of pairing of 3d electrons which leaves two, one and zero unpaired electrons respectively.
The magnetic data agree with maximum spin pairing in many cases, especially with coordination compounds containing d6 ion. However, there are complications with the coordination compounds / species having d4 and d5 ions.
[Mn(CN)6]3– has a magnetic moment equal to two unpaired electrons while [MnCl6]3– has a magnetic moment equal to four unpaired electrons.
Similarly [Fe(CN)6]3– has magnetic moment of a single unpaired electron while [FeF6]3– has a magnetic moment of five unpaired electrons. [CoF6]3– is paramagnetic with four unpaired electrons while [Co(C2O4)]3– is diamagnetic.
This anomalous behaviour is explained by valence bond theory in terms of formation of inner orbitals and outer orbitals complexes.
[Mn(CN)6]3– , [Fe(CN)6]3– and [Co(C2O4)2]3– are inner orbital complexes involving d2sp3 hybridisation, the former two are paramagnetic and the latter diamagnetic. [MnCl6]3– , [FeF6]3– and [CoF6]3– are outer orbital complexes involving sp3d2 hybridisation and are paramagnetic having four, five and four electrons respectively.
7. Crystal Field Theory
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Crystal Field Theory :
The drawbacks of VBT of coordination compounds are, to a considerable extent, removed by the Crystal Field Theory.
The crystal field theory (CFT) is an electrostatic model which considers the metal-ligand bond to be ionic arising purely from electrostatic interaction between the metal ion and the ligand. Ligands are treated as point charges in case of anions or dipoles in case of neutral molecules. The five d orbitals is an isolated gaseous metal atom/ion have same energy, i.e., they are degenerate. This degeneracy is maintained if a spherically symmetrical field of negative charges surrounds the metal atom/ion. However, when this negative field is due to ligands (either anions or the negative ends of dipolar molecules like NH3 and H2O) in a complex, it becomes asymmetrical and the degeneracy of the d orbitals is lost. It results in splitting of the d orbitals. The pattern of spitting depends upon the nature of the crystals field.
(a) Crystal field splitting in octahedral coordination entities :
In an octahedral coordination entity with six ligands surrounding the metal atom/ion, there will be repulsion between the electrons in metal d orbitals and the electrons (or negative charges) of the ligands. Such a repulsion is more when the metal d orbitals is directed towards the ligand than when it is away from the ligand. Thus, the ![]() and
and ![]() orbitals (axial orbitals) which point towards the axis along the direction of the ligand will experience more repulsion and will be raised in energy ; and the dxy , dyz and dzx orbitals (non-axial) orbitals which are directed between the axis will be lowered in energy relative to the average energy in the spherical crystal field. Thus, the degeneracy of the d orbitals has been removed due to ligand electron-metal electron repulsions in the octahedral complex to yield three orbitals of lower energy, t2g set and two orbitals of higher energy, eg set. This splitting of the degenerate levels due to the presence of ligands in a definite geometry is termed as crystal field splitting and the energy separation is denoted by D0 (the subscript o is for octahedral). Thus, the energy of the two eg orbitals will increase by (3/5)D0 and that of the three t2g will decrease by (2/5) D0.
orbitals (axial orbitals) which point towards the axis along the direction of the ligand will experience more repulsion and will be raised in energy ; and the dxy , dyz and dzx orbitals (non-axial) orbitals which are directed between the axis will be lowered in energy relative to the average energy in the spherical crystal field. Thus, the degeneracy of the d orbitals has been removed due to ligand electron-metal electron repulsions in the octahedral complex to yield three orbitals of lower energy, t2g set and two orbitals of higher energy, eg set. This splitting of the degenerate levels due to the presence of ligands in a definite geometry is termed as crystal field splitting and the energy separation is denoted by D0 (the subscript o is for octahedral). Thus, the energy of the two eg orbitals will increase by (3/5)D0 and that of the three t2g will decrease by (2/5) D0.
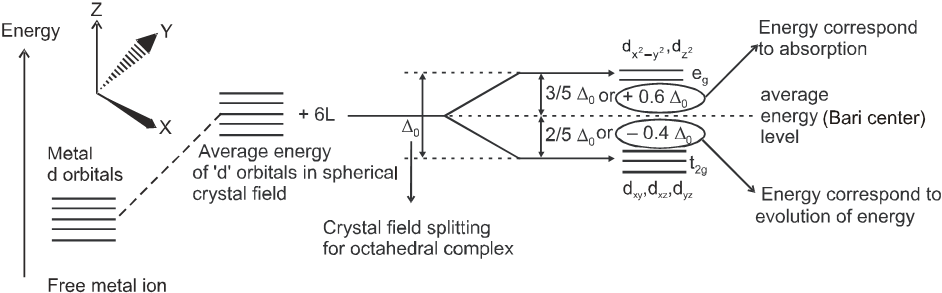
The crystal field splitting, D0, depends upon the fields produced by the ligand and charge on the metal ion. Some ligands are able to produces strong fields in which case, the splitting will be large whereas others produce weak fields and consequently result in small splitting of d orbitals. In general, ligands can be arranged in a series in the orders of increasing field strength as given below :
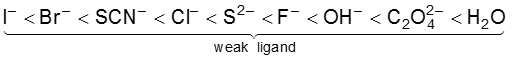 <
< 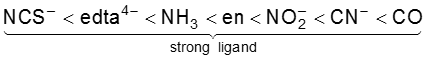
Such a series is termed as spectrochemical series. It is an experimentally determined series based on the absorption of light by complexes with different ligands. For d4 configuration, the fourth electron will singly occupy eg orbital (according to Hund’s rule) or will undergo pairing in t2g orbital, which of these possibilities occurs, depends on the relative magnitude of the crystal field splitting, D0 and the pairing energy, P (P represents the energy required for electron pairing in a single orbital). The two possibilites are :
(i) If D0 < P, the fourth electron enters one of the eg orbitals giving the configuration t32geg1. Ligands for which D0 < P are known as weak field ligands and form high spin complexes.
(ii) If D0 > P, it becomes more energetically favourable for the fourth electron to occupy a t2g orbital with configuration t2g4 eg0. Ligands which produce this effect are known as strong field ligands and form low spin complexes.
(b) Crystal field splitting in tetrahedral coordination entities :
In tetrahedral coordination entity formation, the d orbital splitting is inverted and is smaller as compared to the octahedral field splitting. For the same metal, the same ligands and metal-ligand distances, it can be shown that Dt = (4/9)D0. This may attributes to the following two reasons.
(i) there are only four ligands instead of six, so the ligand field is only two thirds the size ; as the ligand field spliting is also the two thirds the size and
(ii) the direction of the orbitals does not concide with the direction of the ligands. This reduces the crystal field spliting by roughly further two third. So Dt = ![]() ×
×![]() =
= ![]() Do.
Do.
Consequently, the orbital splitting energies are not sufficiently large for forcing pairing and, therefore, low spin configurations are rarely observed.
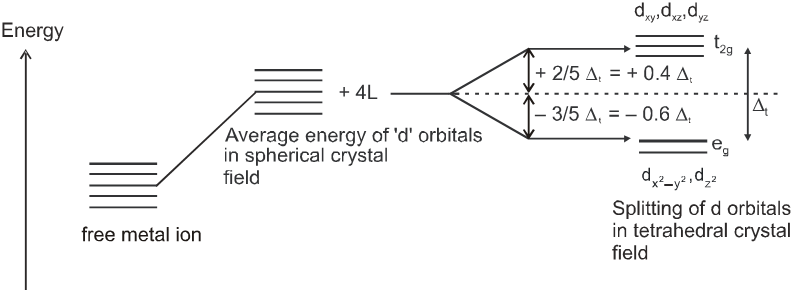
Since Dt < Do, crystal field spliting favours the formation of octahedral complexes.
8. COLOUR IN COORDINATION COMPOUNDS
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Colour in Coordination Compounds :
According to the crystal field theory the colour is due to the d-d transition of electron under the influence of ligands. The mechanism of light absorption in coordination compounds is that photons of appropriate energy can excite the coordination entity from its ground state to an excited state. Consider the Ti(III) ion in solution, that is [Ti(H2O)6]3+. This is a violet colour octahedral complex, where in the ground state of the complex a single electron is present in t2g level. The next higher state available for the transition is the empty eg level. If the light corresponding to the energy of yellow-green is absorbed by the complex, it would excite the electron from t2g level to eg level. Consequently the complex appears violet in colour.
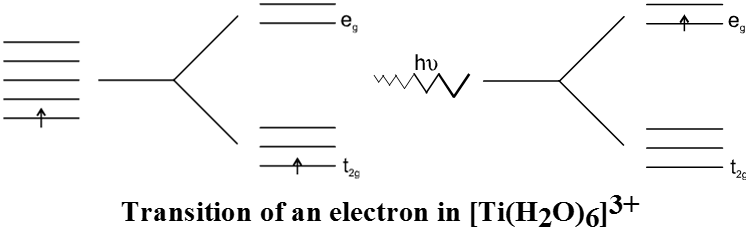
Table below gives the relationship of the wavelength absorbed and the colour observed.
Relationship between the wavelength of light absorbed and the colour observed In some coordination entitles
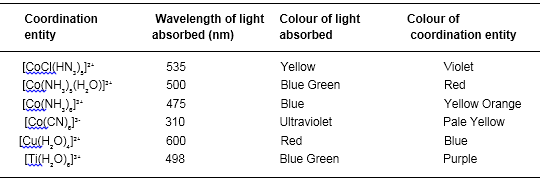
Note :
(a) In absence of ligand, crystal field splitting does not occur and as a consequence the substance appears colourless. For example (i) removal of water from violet coloured complex [Ti(H2O)6]Cl3 on heating makes it colourless. (ii) Similarly anhydrous copper sulphate (CuSO4) is white, but hydrated copper sulphate (CuSO4.5H2O) is blue coloured.
(b) The nature of the ligand and the molar ratio of metal : ligand also influence the colour of the complex for example in the pale green complex of [Ni(H2O)6], the colour change is absorbed when ethylenediamine is progressively added to it.
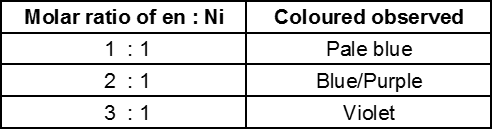
Note : Ruby is Al2O3 in which 0.5–1% Cr3+ ions (d3 electron system) are randomly distributed in the positions normally occupied by Al3+. We may consider Cr(III) species as octahedral Cr(III) complexes incorporated into the alumina lattice ; d-d transition of electron at these centres give rise to the colour (red).
Emerland is the mineral beryl (Be3Al2Si6O18) in which Cr3+ ions occupy octahedral sites, but in this case low energy corresponding to yellow red and blue is absorbed and light corresponding to green region is transmitted.
Limitations of crystal field theory
(1) It considers only the metal ion d-orbitals and gives no consideration at all to other metal orbitals (such as s, px, py and pz orbitals).
(2) It is unable to account satisfactorily for the relative strengths of ligands. For example it gives no explanation as to why H2O is a stronger ligand than OH– in the spectrochemical series.
(3) Account to this theory, the bond between the metal and ligands are purely ionic. It gives no account on the partly covalent nature of the metal ligand bonds.
(4) The CFT cannot account for the p-bonding in complexes.
2. Types of Reagents
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Types of Reagents :
Reagents are of two types :
(i) Electrophiles
(ii) Nucleophiles
Electrophiles :
Electrophiles are electron deficient species.
Ex. ![]() (positively charged species),
(positively charged species),
![]() (species with vacant orbital at central atom).
(species with vacant orbital at central atom).
Nucleophiles and their nucleophilicity :
Nucleophile is a species having negative charge or lone pair of electrons.
They are electron rich species.
Ex. ![]() (l.p on O-atom),
(l.p on O-atom), ![]() (negaively charged species)
(negaively charged species)
Note : :CCl2 is not a nucleophile because it is electron deficient species and act as electrophile.
- Negative ions have more nucleophilic than their neutral species
![]()
- Down the group nucleophilicity increases because the more polarizable donar atom is better nucleophyle
Polarizability size of donar atom
![]()
- Across the period nucleophilicity decreases
![]()
- Bulky base has less nucleophilic character.
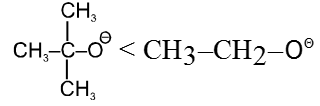
- Effect of solvent : In case of polar aprotic solvents nucleophilicity order of halides is just reversed.
Bases and their basicity :
Bases are the species which accept the proton or which donates l.p. of electron to proton.
- Basicity decreases down the group while nucleophilicity increases
F– > Cl– > Br– > I–
- Nucleophilicity and basicity order will be same across the period.
- For the same donor atom nucleophilicity and basicity order will be same
Leaving group ability :
- Weaker base is better leaving group.
- More resonance stabilised ion will be better leaving group.
- Weaker the carbon-leaving group bond (C–X) better will be the leaving group.
- If activation energy of a reaction is low then reaction will be fast and leaving group will be better.
Ex. (a) I– > Br– > Cl– > F–
(b) CF3SO3– > RCOO– > C6H5O– > OH– >
(c) 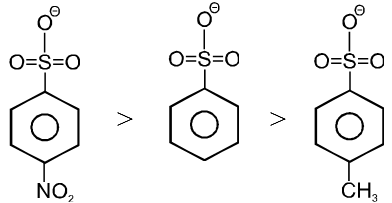
Note : More stable anions are weak bases & hence better leaving group.
3. Types of Solvents
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Types of solvents
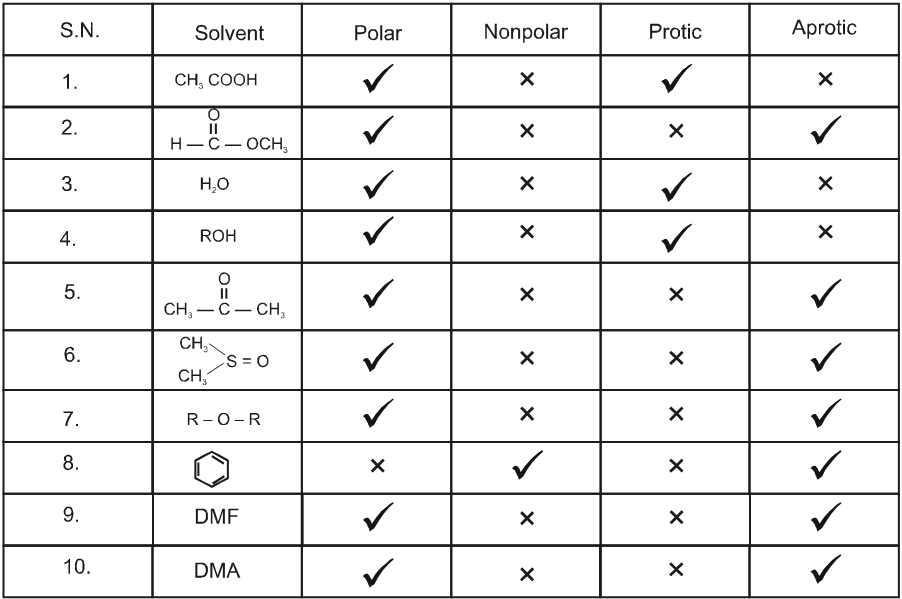
Note : If H atom is attached to oxgyen, nitrogen or sulphur then it is said to be protic solvent.
4. Nucleophilic substitution reactions of alkyl halides
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Nucleophilic substitution reactions of alkyl halides
Alkyl halide undergoes nucleophilic substitution reaction.
![]()
Nucleophilic substitution reactions are of two types :
Unimolecular nucleophilic substitution reaction (SN1) :
- It is a two step process.
- First step is the heterolytic cleavage of carbon halogen bond (
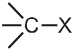 ) to give carbocation intermediate.
) to give carbocation intermediate.
![]() (Ist step is r.d.s.)
(Ist step is r.d.s.)
- In the second step nucleophile attacks from either side of carbocation to generate product (racemic mixture).
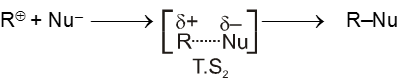 (IInd is fast step)
(IInd is fast step)
Mechanism :
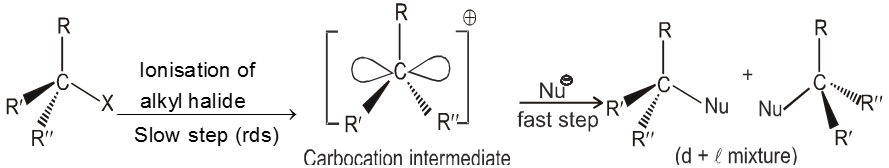
- Carbocation intermediate is formed so rearrangement is possible in SN1 reaction.
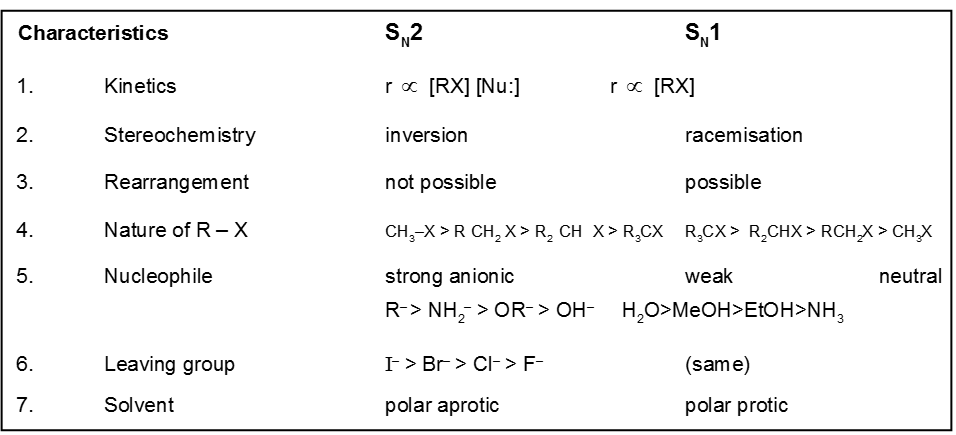
Kinetics :
- Rateµ [Alkyl halide]
- It is unimolecular and first order reaction.
- Rate of SN1 reaction is independent of the concentration and reactivity of nucleophile.
Stereochemistry :
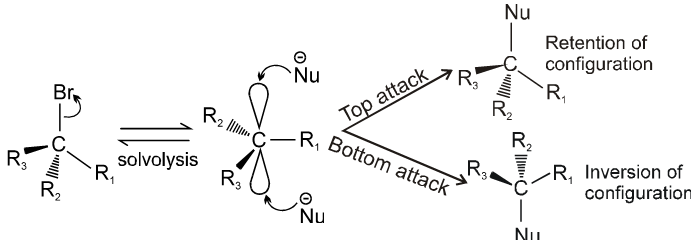
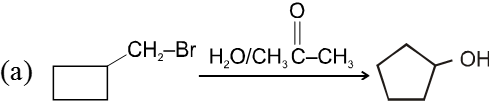
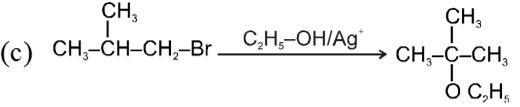
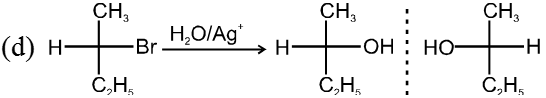
- Catalyst used is Ag+.
- More polar protic solvent is more favourable for SN1.
H2O > ROH > NH3 (order of polar protic solvent).
- In SN1 reaction carbocation is formed along with anion and to solvate these ions, polar protic solvent is used.
- Decreasing order of reactivity of alkyl halides. ;
R–I > R–Br > R–Cl > R–F
Ex.
![]()
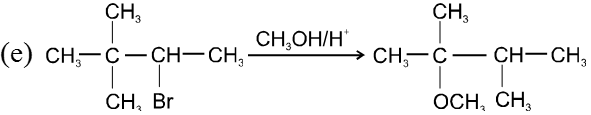
Bimolecular nucleophilic substitution reaction (SN2) :
It is a single step reaction as the rate of reaction depends upon concentration of substrate as well as nucleophile.
Mechanism :
- In this reaction nucleophile attack from back side on the carbon atom bearing leaving group. it is a concerted reaction where bond breaking and bond formation takes place simultaneously to achieve a transition state (trigonal bipyramidal shape) where half bond has been formed and half bond has been broken.
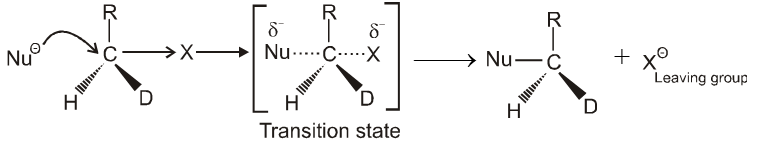
Kinetics :
- rateµ [alkyl halide] [nucleophile]
- It is a bimolecular, one step concerted process.
- It is a second order reaction because in the r.d.s. both species are involved.
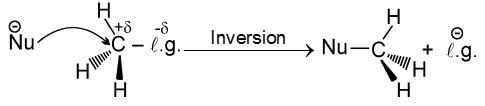
Steriochemistry :
Only one product is formed where inversion of configuration takes place.
- Polar aprotic solvent is favourable not polar protic solvent. Becuase in case of polar protic solvent nucleophilicity of anion is decreased due to solvation and such solvation is not possible in case of polar aprotic solvent.
- Electron withdrawing group increases the rate of sN2 reaction.
O=CH–CH2–Br > CH3–O–CH2–Br > H–CH2–Br > CH3–CH2–Br
Comparision between SN1 / SN2 reaction :
Examples of SN2 reaction of alkyl halides :
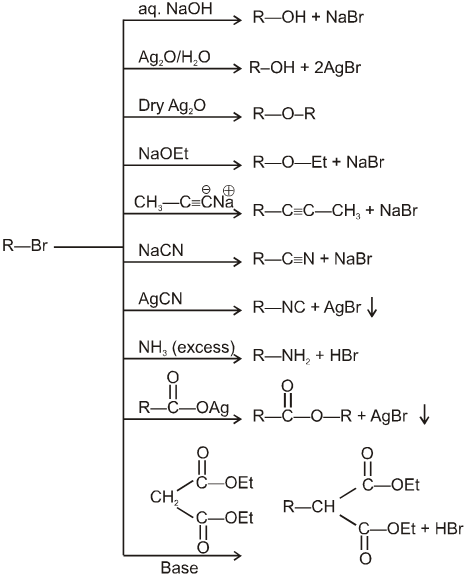
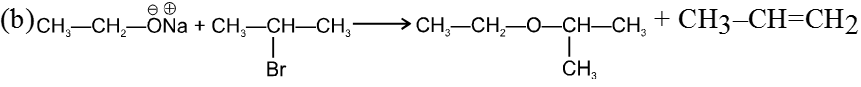
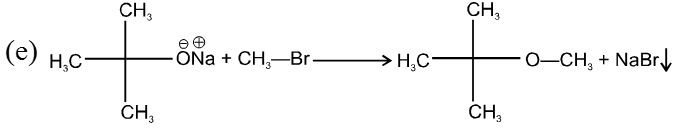
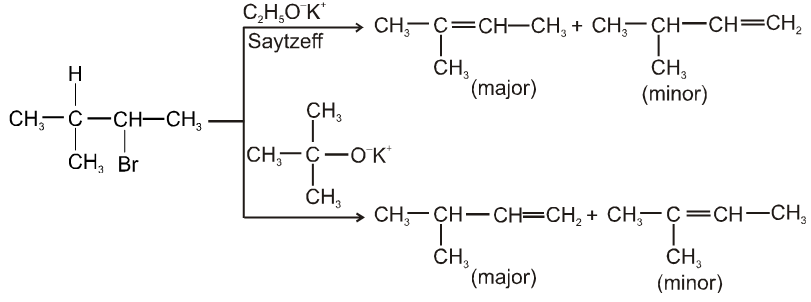
(i) Williamson’s synthesis of ethers :
- It is used for the preparation of symmetrical as well as unsymmetrical ether.
R—Br + NaOEt ¾¾®R—OEt + NaBr
- Williamson’s synthesis involve attack of an alkoxide on alkyl halide to give ethers. In place of alkoxide, phenoxide can also be used.
- This is an SN2 reaction because alkoxide is a strong nucleophile.
- On using 2º & 3º alkylhalide we get alkene not ether as a product.
Ex.
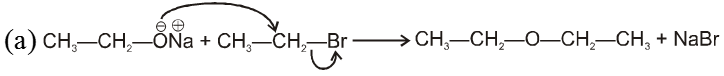
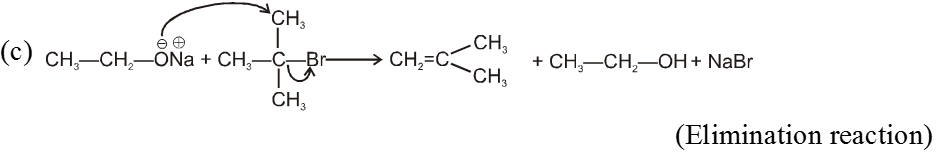
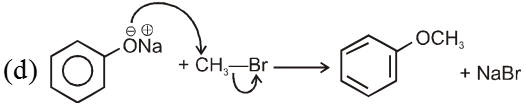
Note : Vinyl or Aryl halide should not be used because they don’t give SN2 reaction.
Ex. 
(ii) Reaction with AgCN and NaCN :
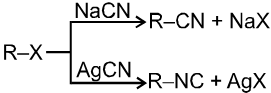
- NaCN is a more ionic hence ionized to give free
 (an ambident nucleophile) where carbon side is more active than nitrogen side. That is why with NaCN, RCN is formed.
(an ambident nucleophile) where carbon side is more active than nitrogen side. That is why with NaCN, RCN is formed.
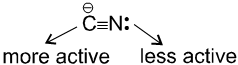
- On the other hand AgCN is more covalent so it is not ionized therefore only nitrogen side is free to act as a nucleophile and give isocyanide (R—NC)
(iii) Reaction with NH3 :

![]()
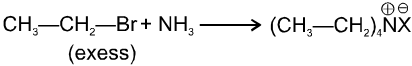
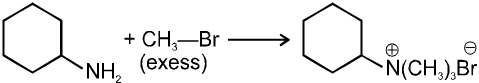
(iv) Finkelstein reaction :
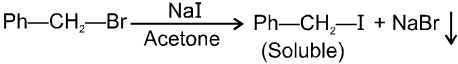
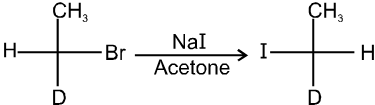
R—OH + HI¾¾®R—I ![]() R—H + I2 (reduced) [Final product is Alkane (R-H) not R-I]
R—H + I2 (reduced) [Final product is Alkane (R-H) not R-I]
This is a problems that is why iodides are best prepared through halogen exchange reaction. It is also known as Finkelstein reaction. In this reaction R—Cl and R—Br is treated with sodium iodide in acetone.
![]()
- NaI is soluble in acetone. In this reaction acetone is used because sodium iodide is soluble in acetone but NaBr and NaCl are insoluble so precipitated out. This eliminates any possibility of reverse reaction.
- It is an SN2 reaction therefore only 1ºRX and 2ºRX is used.
5. Aryl Halides
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Aryl Halides
Preparation of Aryl Halides
1. By Halogenation :
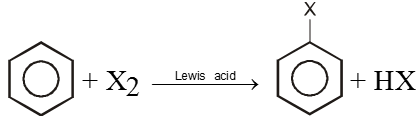
X2 = Cl2, Br2 ; Lewis acid = FeCl3, AlCl3, ZnCl2, etc.
2. By Decarboxylation :
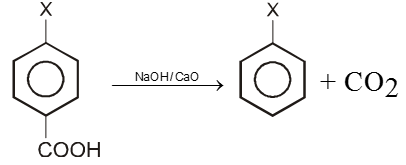
Chemical reaction of Aryl halide (Bimolecular nucleophilic substitution SN2 Ar)
- An electron withdrawing group at ortho or para positions with respect to a good leaving groups are necessary conditions for SN2 Ar.
Mechanism :
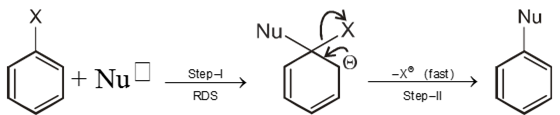
Intermediate ion is stabilized by resonance and are stable salts called Meisenheimer salts.
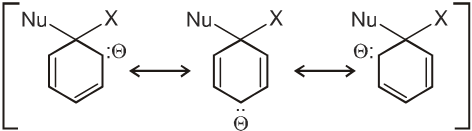
- A group that withdraws electrons tends to neutralize the negative charge of the ring and this dispersal of the charge stabilizes the carbanion.
G withdraws electrons : stabilizes carbanion, activates the Ar-SN2 reaction.
(–![]() (CH3)3, –NO2, –CN, –SO3H, –COOH, –CHO, –COR, –X)
(CH3)3, –NO2, –CN, –SO3H, –COOH, –CHO, –COR, –X)
- A group that releases electrons tends to intensify the negative charge, destabilizes the carbanion, and thus slows down reaction.
G (–NH2, –OH, –OR, –R) releases electrons : destabilizes carbanion, deactivates the Ar-SN2 reaction.
Element effect :- The fact that fluoro is the best leaving group among the halogens in most ArSN2 but in SN1 & SN2 mechanism where fluoro is the poorest leaving group among halogens.
6. Elimination reactions of alkyl halides
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
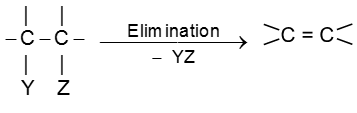
Elimination reactions of alkyl halides
In an elimination reaction two atoms or groups (YZ) are removed from the substrate and generally resulting into formation of p bond.
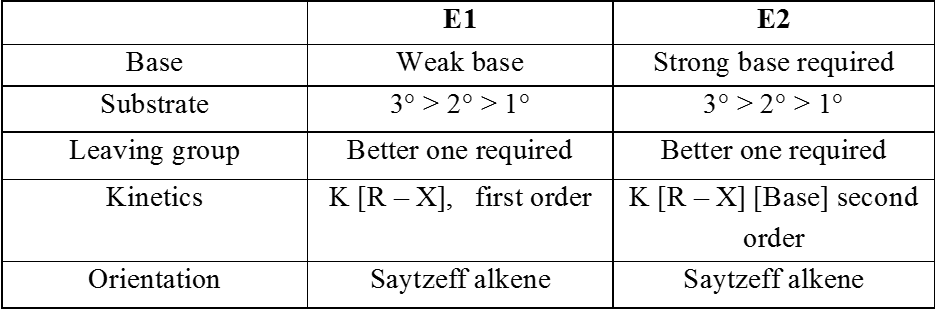
Unimolecular elimination reaction (E1) :
Proton and leaving group depart in two different step.
Mechanism :
Step 1 : Formation of the carbocation (r.d.s.)

Step 2 : Base ( ) abstracts a proton (fast step)

- Reaction intermediate is carbocation, so rearrangment is possible
Kinetics:
- Rateµ[Alkylhalide]
- It is a unimolecular and first order reaction.
- Reactivity order is similar to SN1 becuase Carbocation Intermediate is formed in the rds step.
SN1 v/s E1 :
- In case of alkyl halides SN1 product is generally more than E1 product
Ex.
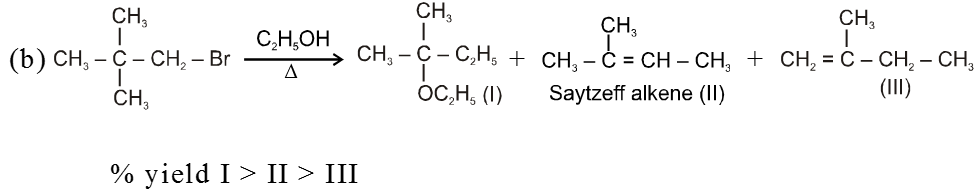
Bimolecular elimination reaction (E2) :
- It is a second order reaction because rate of reaction depends upon conc. of substrate as well as base.
Mechanism :
- Orientation of eliminated proton and leaving group are antiparallel or antiperiplanar to each other because in anti conformation the transition state is more stable due to minimum electronic repulsion.
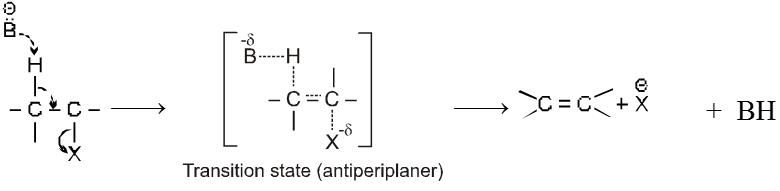
- E2 reaction is stereospecific.
- E2 reaction also depends upon size of base which decides major or minor product.
Kinetics :
- Rate µ [R – X] [Base]
- This is a single step, bimolecular reaction
- No carbocation intermediate is formed hence there is no rearrangement but a transition state is achieved because it is a single step reaction.
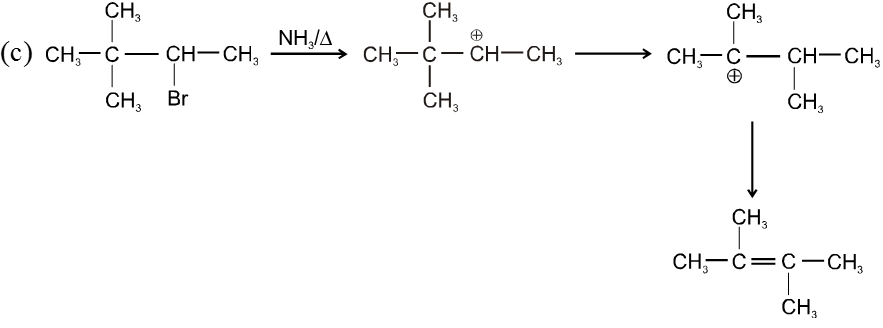
- More favourable substrate is tertiary alkyl halide because it will give more stable alkene according to saytzeff rule.
Ex.
Some important terms :
(a) Regioselective reaction : Those reaction in which more than one structural isomeric products are possible, said to be regioselective reaction.
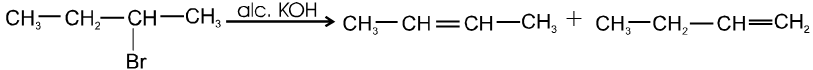
(b) Regiospecific reaction : Those reaction in which only one structural isomer product is formed out of the possible products, said to be regiospecific reaction.
(c) Stereoselective : Those reactions in which mixture of two stereoisomeric products are formed with one major product.
Examples : SN1, SN2 and E2
(d) Stereospecific reaction : Those reactions in which two stereoisomeric reactants give two different stereoisomeric products, are called as stereospecific reactions.
Examples : SN2 and E2
Note : All stereospecific reactions are stereo selective.
Comparison between E1 and E2 reactions :
E1cB Reaction :
- It is two step reaction.
- In first step base takes proton from adjacent carbon atom of halogen bearing carbon and generate carbanion intermediate.
-
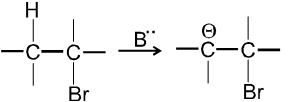
- In second step there is loss of leaving group by carbanion to get alkene :
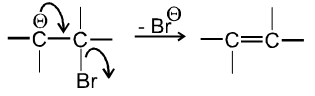
Ex.
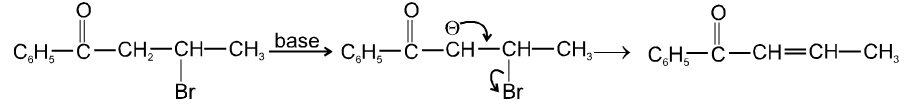
- b-hydrogen atom should be more acidic which is possible only if carbon atom having b-hydrogen atom should be link to with electron withdrawing group (-m, -I group).
- Leaving group should be more electronegative because it also increases acidity of b-hydrogen atom.
- Experimentally it is found that Ist step i.e. formation of carbanion intermediate is fast step and second step i.e. removal of leaving group is slow step thus r.d.s. in E1cB is second step.
7. Reactions of chloroform
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Reactions of chloroform
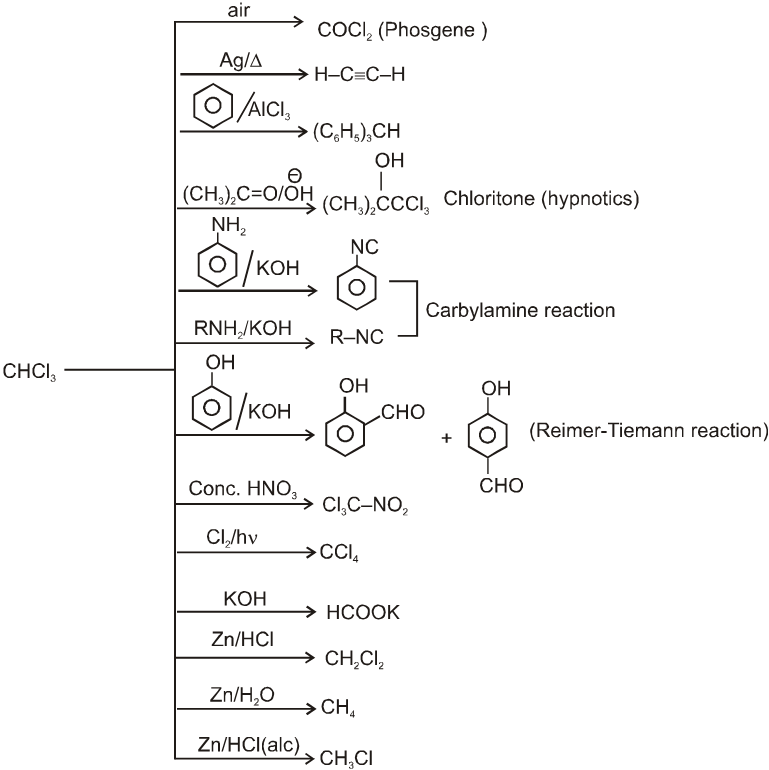
- Purity of chloroform (presence of phosgene) can be tested before use as anaesthetic by treating with aqueous solution of AgNO3 because the presence of COCl2 may cause cardiac failure.
- Chloroform is stored in dark colour bottle containing small amount of ethyl alcohol. (It converts phosgene into diethylcarbonate).
2. Preparation methods of Aldehydes and Ketones
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Preparation methods of Aldehydes and Ketones
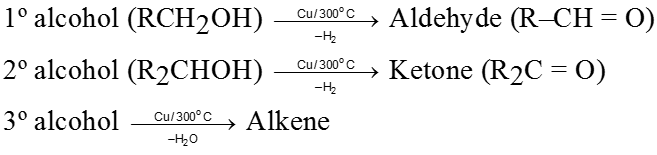
By oxidation of alcohols :
Primary alcohols ![]() Aldehydes
Aldehydes
Secondary alcohols ![]() Ketones
Ketones
By dehydrogenation of alcohols :
Dehydrogenation means removal of hydrogen and reagent used is heated copper.
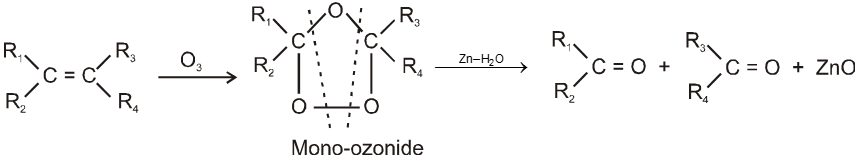
Ozonolysis of alkene :
It is used to get carbonyl compounds from alkene. The reaction is
Note :
(i) During the cleavage of ozonide Zn is used to check further oxidation of aldehyde into acid.
(ii) By this method we can locate double bond in olefin or exact structrue of hydrocarbon can be determined by knowing ozonolysis product i.e. by placing double bond at the place of two carbonyl oxygen atoms of two carbonyl compounds.
(iii) Among the three molecules of carbonyl compounds.
(a) If one molecule contains two carbonyl groups, then hydrocarbon will be alkadiene.
(b) If all the three molecules contain two carbonyl group then hydrocarbon will be cycloalkatriene.
Wacker process :
Alkenes can directly be oxidised to corresponding aldehydes or ketones by treating them with a solution of PdCl2 containing a catalytic amount of CuCl2 in presence of air or O2 . Except ethene any higher alkene will give ketone.
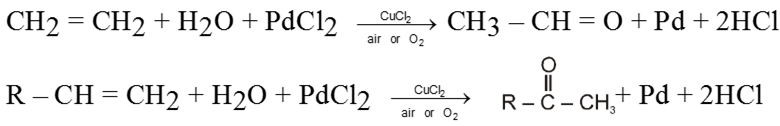
Note : During the reaction PdCl2 is reduced to Pd and CuCl2 is reduced to Cu(I)
Hydration of alkynes :
![]()
Other alkynes give ketones in this reaction.
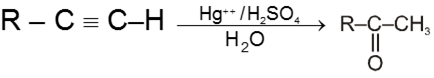
Hydroboration of alkyne :
It is used to get aldehyde from terminal alkyne. Here reagent is (i) diborane (B2H6) (ii) H2O2,OH–
![]()
In this reaction Borane (BH3) is act as electrophile.
Dry distillation of calcium salt of acid :
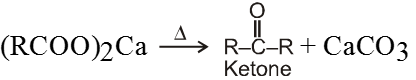
Ex.
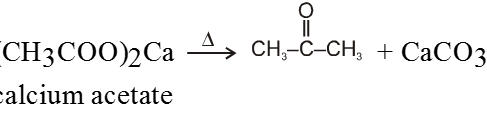
On dry distillation of calcium salt of acetic acid with calcium salt of formic acid we get a mixture of aldehyde, ketone and formaldehyde.
On passing vapours of fatty acids over Mangnous oxide at 300ºC :
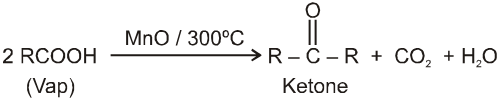
On passing mixture of vapours of fatty acid with formic acid we get a mixture of aldehyde, ketone and formaldehyde.
On aqueous alkali hydrolysis of gem-dihalides :
Terminal gemdihalides will give aldehyde while non-terminal will give ketone as follows
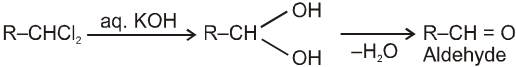
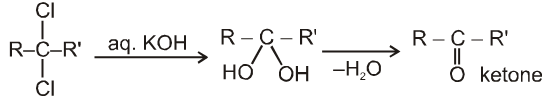
3. Methods used for the preparation of Aldehydes only
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Methods used for the preparation of Aldehydes only
Rosenmund's reduction :
Here acid chlorides are reduced to aldehyde with H2 in boiling xylene using palladium as a catalyst supported on barium sulphate.
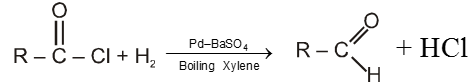
Note :
(a) Pd Catalyst is poisoned by BaSO4 to check further reduction of aldehyde to alcohol.
(b) Formaldehyde cannot be obtained by this method because HCOCl is unstable at common temperature.
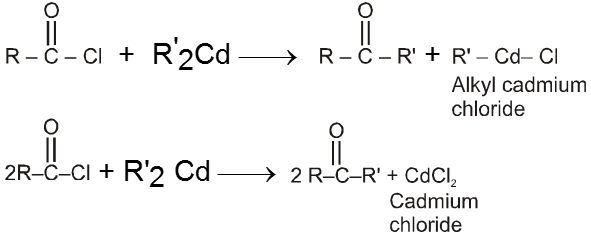
(c) Reaction with acid chloride and dialkyl cadmium we can obtain ketone.
Stephen's reduction :
![]()
Oxo-process :
It is also called as carbonylation here alkene reacts with water gas at high temperature and pressure in the presence of cobalt carbonyl catalyst to give aldehyde.
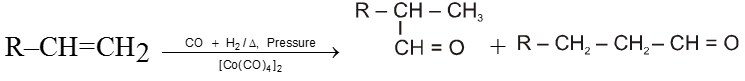
Reimer-Teimann Reaction :
By this method phenolic aldehyde is prepared

From esters or nitrile :
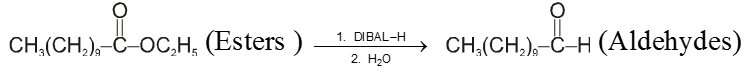
![]()
DIBAL–H : Diisobutyl aluminium hydride [AlH(i-Bu)2] is a reducing agent.
From hydrocarbons :
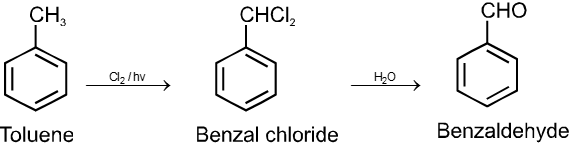
By oxidation of methyl benzene and its derivative using chromyl chloride (CrO2Cl2)
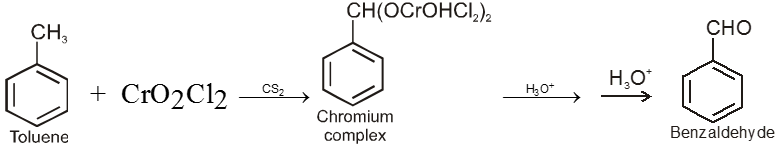
This reaction is called Etard reaction.
By oxidation of methyl benzene and its derivative using chromic oxide (CrO3) in acetic anhydride:

By Gatterman-Koch reaction :
![]()
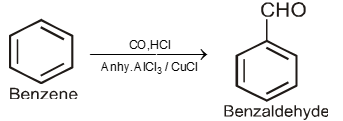
4. Methods used for the preparation of Ketones only
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Methods used for the preparation of Ketones only
Using alkanoylchloride and dialkyl cadmium :
By acylation or benzoylation of aromatic hydrocarbon (Friedel-Craft Reaction)
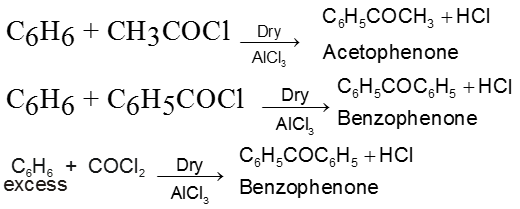
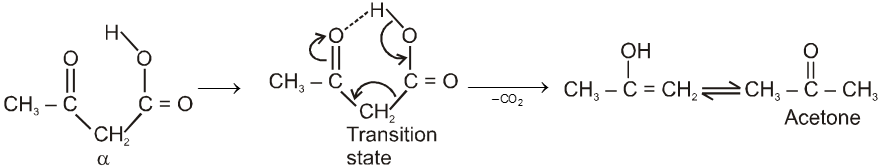
By acid hydrolysis followed by heating of b-Ketoester :
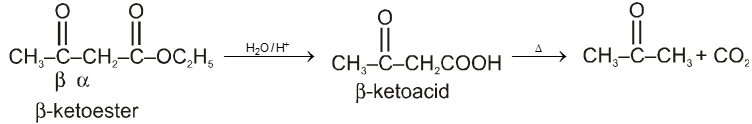
Note : It is b-ketoacid which decarboxylate more readily as it proceeds via six membered cyclic transition-state.
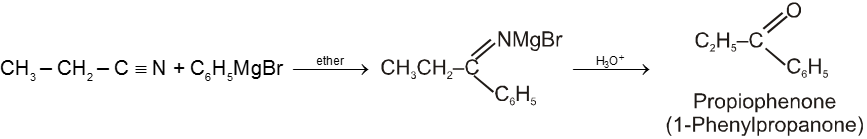
From nitriles :
Treatment of nitrile with Grignard reagent followed by hydrolysis gives a ketone.

5. Physical properties of Aldehydes and Ketones
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Physical properties of Aldehydes and Ketones
Physical properties of Aldehydes and Ketones :
- Methanal - Gas at room temperature
- Ethanal - Volatile liquid
- Other aldehydes and ketones - Liquid or solid at room temperature
- Boling points of aldehydes and ketones are higher than those of hydrocarbons and ethers of comparable molecular masses.
Reason : Weak molecular association in aldehydes and ketones, arising out of the dipole-dipole interactions
- Boiling points of aldehydes and ketones are lower than those of alcohols of similar molecular masses.
Reason : Absence of intermolecular hydrogen bonding
- Lower members of aldehydes and ketones are miscible with water in all proportions.
Reason : They form hydrogen bonds with water.
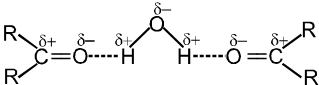
- Solubility of aldehydes and ketones decreases rapidly on increasing the length of the alkyl chain.
- All aldehydes and ketones are fairly soluble in organic solvents such as ether, methanol, etc.
- Lower aldehydes have sharp pungent odours.
- As the size of aldehydes increases, the odour becomes less pungent and more fragrant.
6. Chemical Reactions
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chemical Reactions
Nucleophilic addition reactions :
Addition of a nuceophile and a proton across the (C = O) double bond. The reactivity of the carbonyl group arises from the electronegativity of the oxygen atom and the resulting polarization of the carbon-oxygen double bond. The electrophilic carbonyl carbon atom is sp2 hybridized and flat, leaving it relatively unhindered and open to attack from either face of the double bond.
Mechanism :
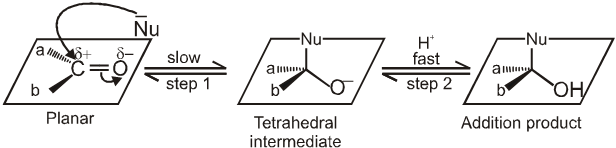
Nucleophile (Nu-) attacks the carbonyl group perpendicular to the plane of sp2 hybridised orbitals of carbonyl carbon.
In the process, hybridisation of carbon changes from sp2 to sp3.
A tetrahedral alkoxide is formed as intermediate.
Reactivity : Aldehydes are more reactive than ketones in nucleophilic addition reactions.
![]()
There are two factors which influence the reactivity of ketone and aldehyde.
(i) Inductive effect (ii) steric factor
(i) + I effect of alkyl group decrease the amount of charge on C+ (C+ – O–). in ketones.
ii) Steric effect also causes the less reactivity of carbonyl group.
(I) Addition of hydrogen cyanide (HCN)
![]()
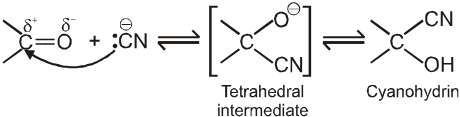
Note :
(i) Addition of HCN over aldehyde and ketones gives cyanohydrin.
(ii) Cyanohydrin on acid hydrolysis gives a-hydroxy acid.
(iii) Cyanohydrin on treating with NH3(l) followed by acid hydrolysis gives a-amino acid.
(iv) In case of ketone cyanohydrin formation is reversible due to bulky group of ketone which hinder the formation.
(II) Addition of sodium hydrogen sulphite (NaHSO3)
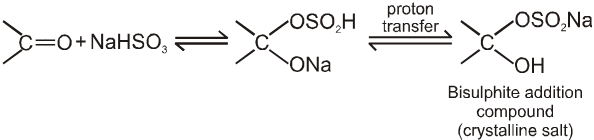
salt on hydrolysis gives carbonyl compounds again, this reaction is used to separate the aldehydes from mixture.
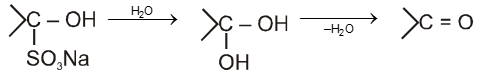
(III) Addition of alcohols (ROH) :
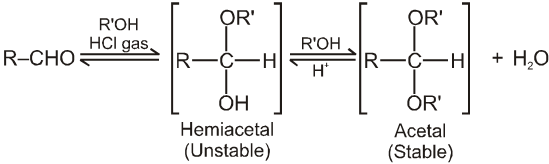
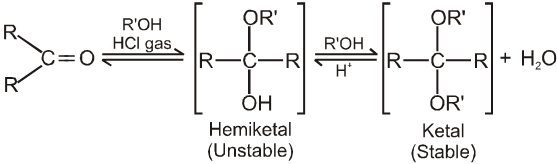
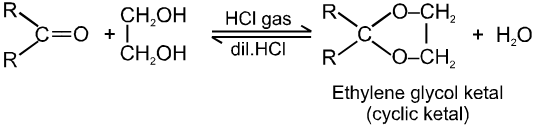
Note :
(i) Acetal is formed to protect aldehyde for a long time.
(ii) Acetal has functional groups ether.
(iii) Acetal formed can be decomposed to original aldehyde by dilute acid.
(iv) On treating with ethyleneglycol we get cyclic acetal or ketal.
(v) Acetal formation is found to be more favourable than ketal formation if both the carbonyl groups are present within the molecule.
(IV) Addition of water :
Aldehyde or ketone reacts with water to form gem-diols. Water is a poor nucelophile and therefore adds relatively slowly to the carbonyl group, but the rate of reaction can be increased by an acid catalyst.
Mechanism:
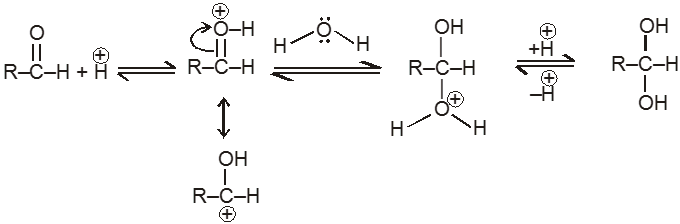
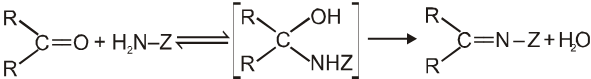
(V) Addition of ammonia and its derivatives (addition elimination reactions) :
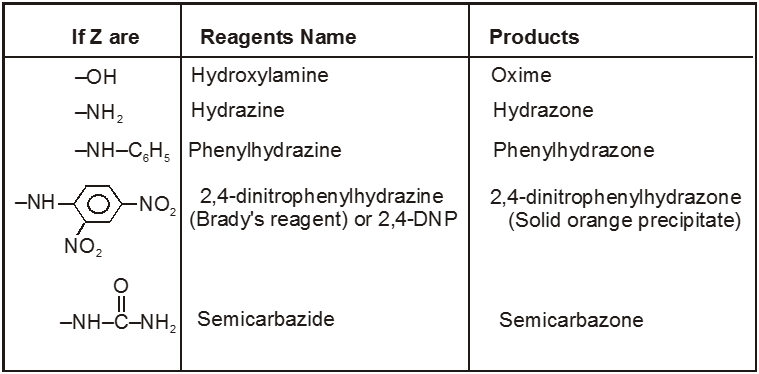
(VI) Addition of Grignard reagents (Preparation of alcohol) :
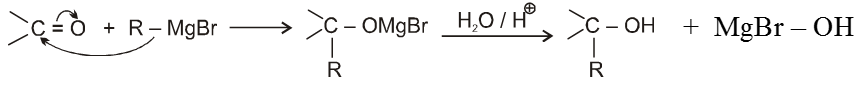
(a) When formaldehyde is treated with Grignard reagent followed by acid hydrolysis primary alcohol is obtained.
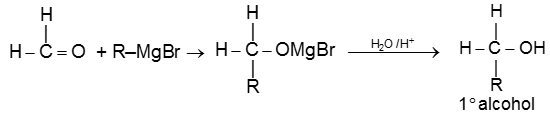
(b) When aldehyde except formaldehyde is treated with grignard reagent followed by hydrolysis 2° alcohol is obtained.
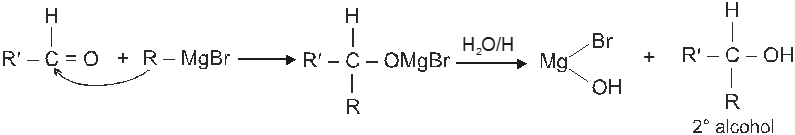
(c) When ketone is treated with grignard reagent followed by acid hydrolysis 3° alcohol is obtained.
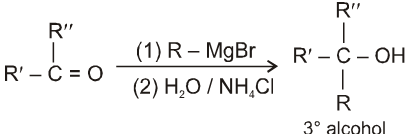
Beckmann rearrangement in Oximes:
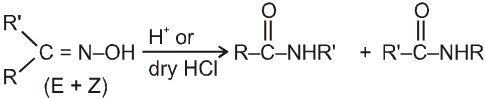 (If R' is bulkier than R)
(If R' is bulkier than R)
Mechanism :
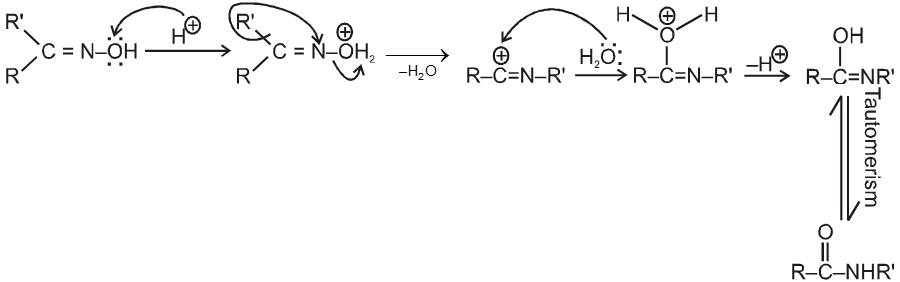
Note :
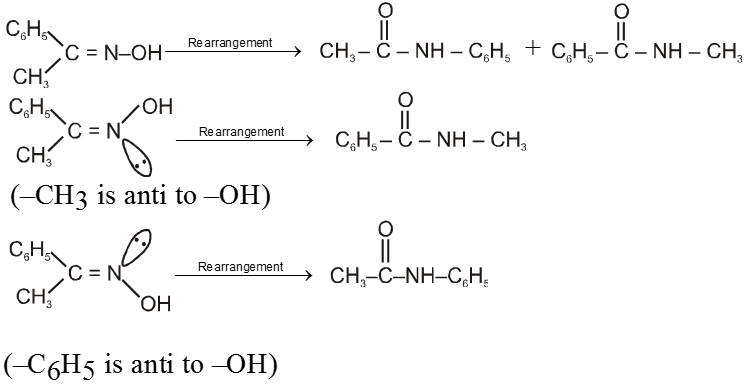
(i) Oxime undergoes Beckmann rearrangement to give its isomer amide.
(ii) In this reaction the group which is anti to –OH group migrates.
Ex.
Reactions due to a-Hydrogen
a-Hydrogen of aldehydes and ketones are acidic: They undergo a number of reactions due to the acidic nature of a-hydrogen.
Reason for the acidity of a-hydrogen: Strong electron-withdrawing effect of the carbonyl group, and resonance stabilisation of the conjugate base
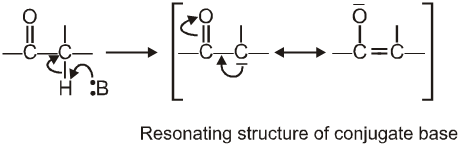
(I) Aldol condensation (or aldol reaction)
Aldehydes and ketones with at least one a-hydrogen undergo a reaction in the presence of dilute alkali as catalyst.
Mechanism
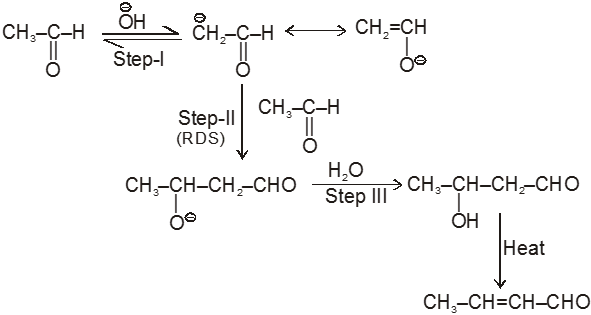
Ex.
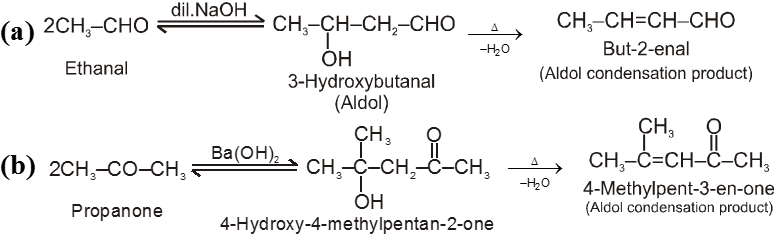
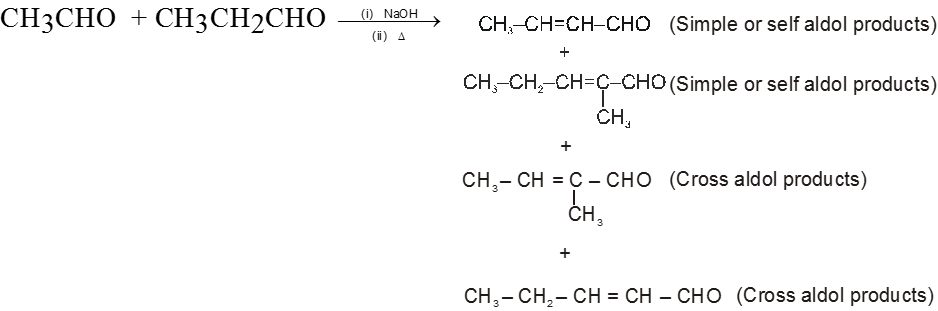
(II) Cross-Aldol condensation :
On using two types of carbonyl compounds both having a-hydrogen atoms we get a mixture of four condensed product because two types of carbonyl compounds will give two type of carbanions which will be nucleophile for itself and other molecule.
Ketones can also be used as one component in cross-aldol reactions.
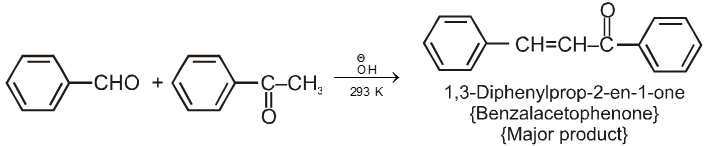
Note : On using formaldehyde and acetaldehyde during crossed aldol all the a-hydrogen atom of acetaldehyde are replaced one by one by hydroxymethyl group because of smaller size of formaldehyde to give trihydroxymethylacetaldehyde which undergoes crossed cannizaro's reaction with formaldehyde to give tetrahydroxymethyl methane and formate ion as a final product.
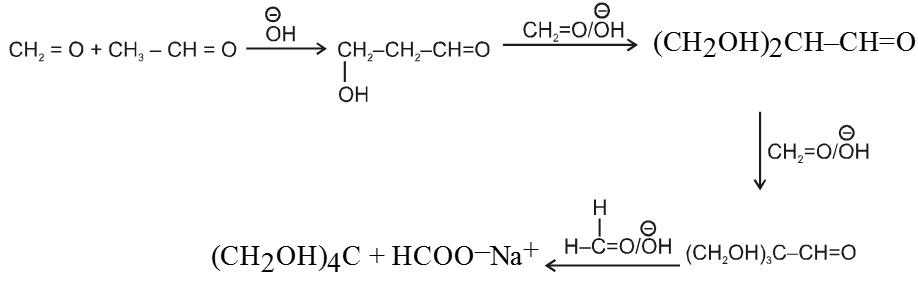
Ex. Show how cinnamaldehyde is prepared by crossed aldol condensation ?
Sol. ![]()
(III) Intramolecular aldol condensation :
If two carbonyl groups with a-hydrogen atoms are present within the same molecule, then we get cyclic a, b-unsaturated aldehyde / ketones via the formation of cyclic-b-hydroxy aldehyde / ketone in presence of basic medium.
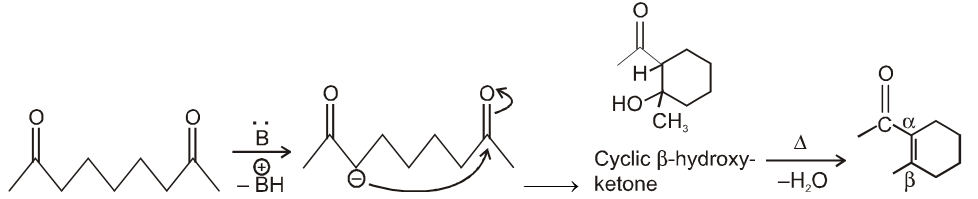
By knowing product we can get reactant as in case of intermolecular aldol condensation :
Perkin reaction :
When aromatic aldehyde like benzaldehyde is treated with anhydride in the presence of sodium salt of acid from which anhydride is derived we get a, b-unsaturated acid.

Mechanism :
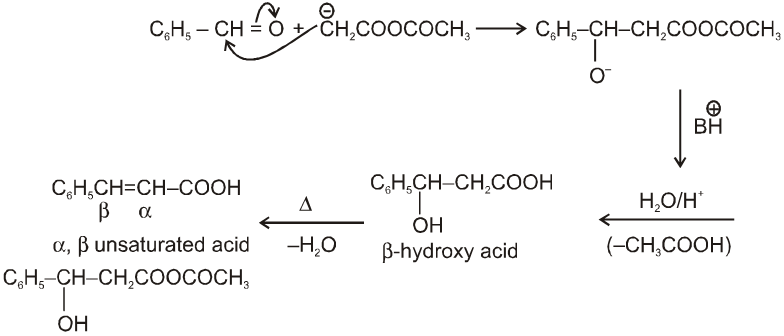
Note : By knowing a, b-unsaturated acid we can get idea about the anhydride used in perkin reaction. This can be done by keeping 'H' at a and –OH at b-carbon atom followed by breaking a, b- carbon. By this we can know about acid and it will be anhydride of this acid only.
Cannizzaro reaction :
Aldehydes which do not have an a- hydrogen atom, undergo self oxidation and reduction (disproportionation) reaction on treatment with a concentrated alkali.
Ex.
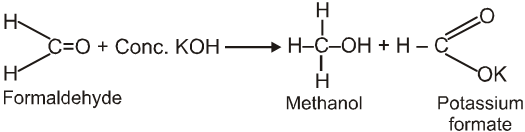
Mechanism :
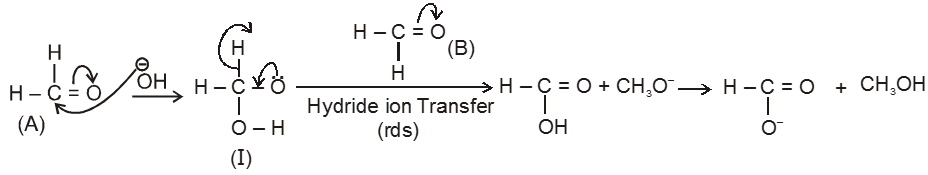
By this mechanism it is clear that acid corresponds to that carbonyl compound over which ![]() can attack easily as nucleophile.
can attack easily as nucleophile.
Note : It is observed that hydride ion transfer from (I) to Carbonyl compound (B) is rate determining step.
Crossed Cannizzaro reaction :
On using two types of carbonyl compounds not having a-hydrogen atom, acid salt will be corresponding to that aldehyde over which ![]() will approach without any hindrence.
will approach without any hindrence.
(i) 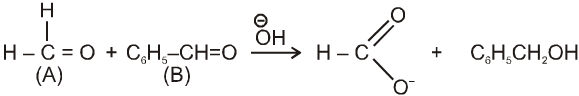
(ii) 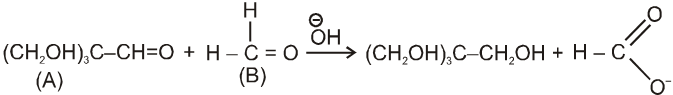
in case (i) ![]() will easily go to (A) and in case (ii) it will go to (B) hence acid salt will be formate ion in both the cases.
will easily go to (A) and in case (ii) it will go to (B) hence acid salt will be formate ion in both the cases.
Intramolecular Cannizzaro reacion :
Here two carbonyl groups (without a-hydrogen atom) are present within the same molecule.
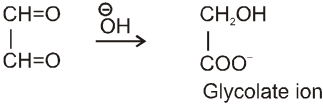
Mechanism :
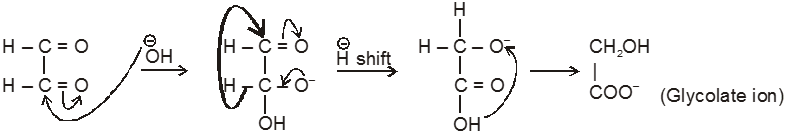
Ex.
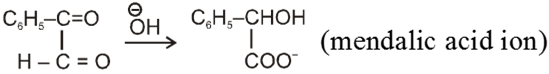
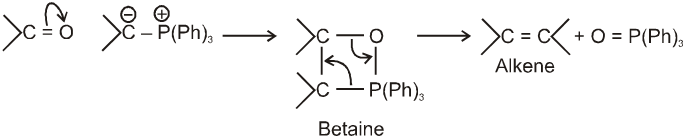
Wittig reaction :
It is used to get alkene from carbonyl compound using phosphorus ylide via the formation of cyclic structure betaine.
Mechanism :
Note : Phosphorus ylides are prepared from alkylhalide and triphenylphosphine in the presence of base like sodium ethoxide as –
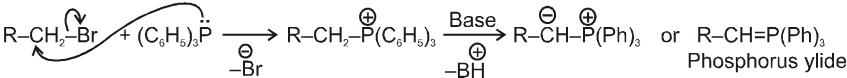
Ex.
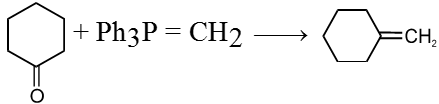
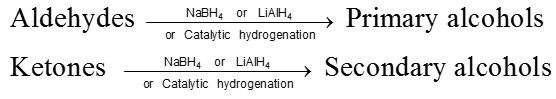
Reduction reactions
(I) Reduction to alcohols :
(II) Clemmensen reduction :
Used to get alkane from carbonyl compounds.
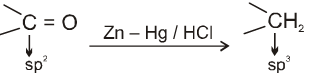
Note : Generally this reaction is avoid if acid sensitive groups are present in the carbonyl compounds.
(III) Wolf-Kishner reduction :
Used to get alkane from carbonyl compounds.
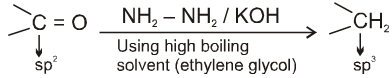
Note : Generally this reaction is avoid if base sensitive groups are present in the carbonyl compounds
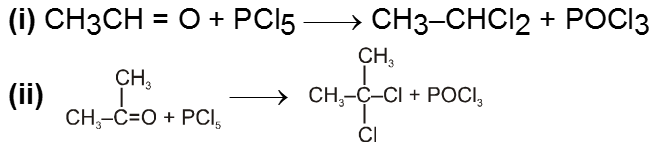
(IV) Reaction with PCl5 :
Carbonyl compounds give gemdihalides
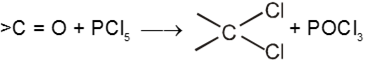
(V) Pinacol-Pinacolone rearrangement :
Pinacole is obtained when 2 moles of acetone are heated with divalent active metal magnesium followed by treating with water.
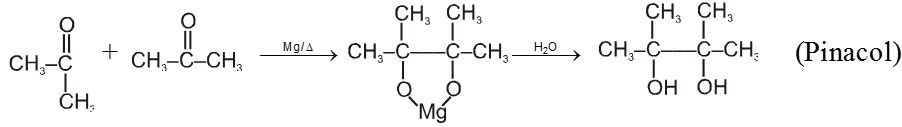
Pinacole undergoes rearrangement in acidic media to give pinacolone
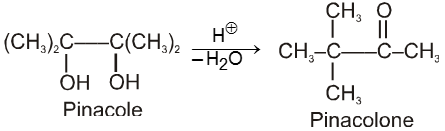
Oxidation reactions
(I) Haloform reaction :
Acetaldehyde and methylalkyl ketones react rapidly with halogen (Cl2, Br2 or I2) in the presence of alkali to give haloform and acid salt.
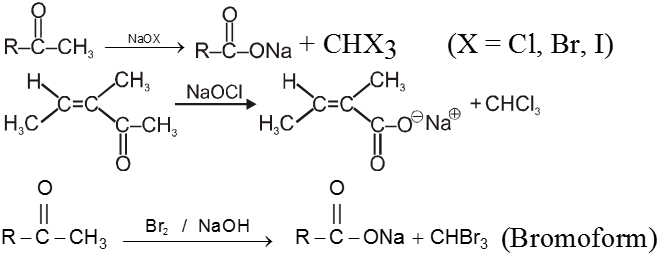
In this reaction – CH3 of  group is converted into haloform as it contains acidic hydrogen atom and rest-part of alkyl methyl ketone give acid salt having carbon atom corresponding to alkyl ketone.
group is converted into haloform as it contains acidic hydrogen atom and rest-part of alkyl methyl ketone give acid salt having carbon atom corresponding to alkyl ketone.
Preparation of haloform from methylketone involves two steps.
(a) Halogenation
(b) Alkalihydrolysis
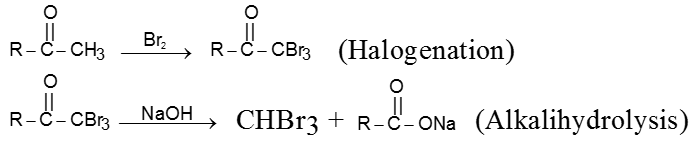
Note : This reaction is used to distinguish the presence of 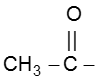 group.
group.
(II) Oxidation of Aldehydes :
• Aldehydes are oxidised to carboxylic acids by common oxidising agents such as KMnO4, HNO3, K2Cr2O7, etc.
![]()
• Aldehydes are also oxidised by mild oxidising agents such as Tollen’s reagent and Fehling’s reagent. On the other hand, ketones are not oxidised by mild oxidising agents.
• Ketones are oxidised under vigorous conditions, i.e., by strong oxidising agents and at elevated temperatures.
It involves carbon-carbon bond cleavage.
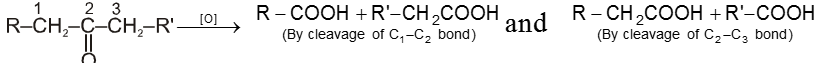
(a) Tollen’s reagent :
It is ammonical silver nitrate solution, prepared by adding ammonium hydroxide to AgNO3 solution. During reaction, first Ag2O is formed which is dissolved in ammoniumhydroxide to give Tollen’s reagent.
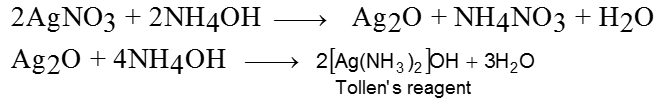
Tollen’s reagent is weak oxidising agent. It gives Ag mirror test with aldehyde.

(b) Fehling’s solution :
It is an alkaline solution of cupric ion complexed with sodium potassium tartarate.
There are two solutions in Fehling solution
Solution (A) CuSO4 solution and
Solution (B) Alkaline solution of sodium potassiumtartarate.
When these two solutions are mixed we get deep blue coloured solution.
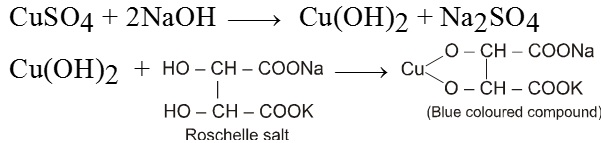
Equal volume of both the solutions are heated with aldehyde to give red brown precipitate of cuprous oxide (Cu2O) which confirms the presence of aldehyde
![]()
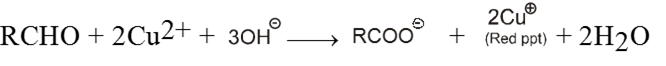
(c) Benedict solution :
It also consists of two solutions.
Solution (A) CuSO4 solution and
Solution (B) Alkaline solution of sodium Citrate.
CuSO4 + 2NaOH Cu(OH)2 + Na2SO4
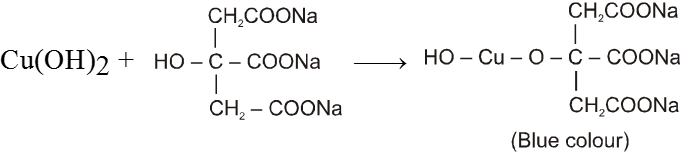
Aldehyde gives positive test with Benedict solution.
![]()
(d) Schiff’s reagent :
It is dilute solution of rosaniline hydrochloride whose pink colour has been discharged by passing SO2. Aldehyde restores pink colour when treated with schiff’s reagent (Magenta solution in H2SO3).
Other miscellaneous reactions :
(I) 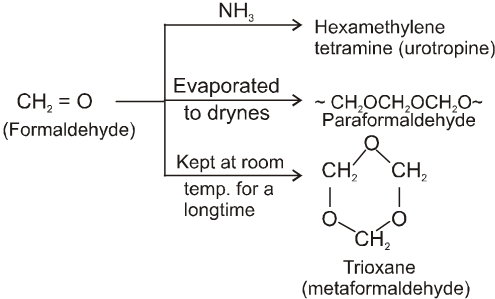
(II) 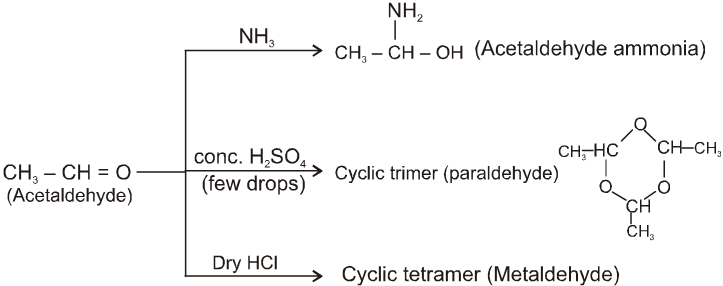
(III) 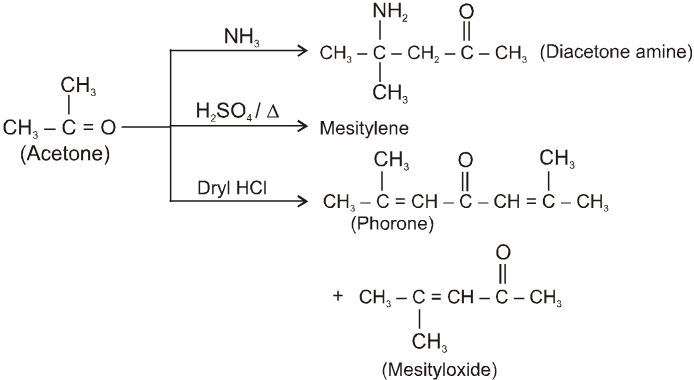
(IV) 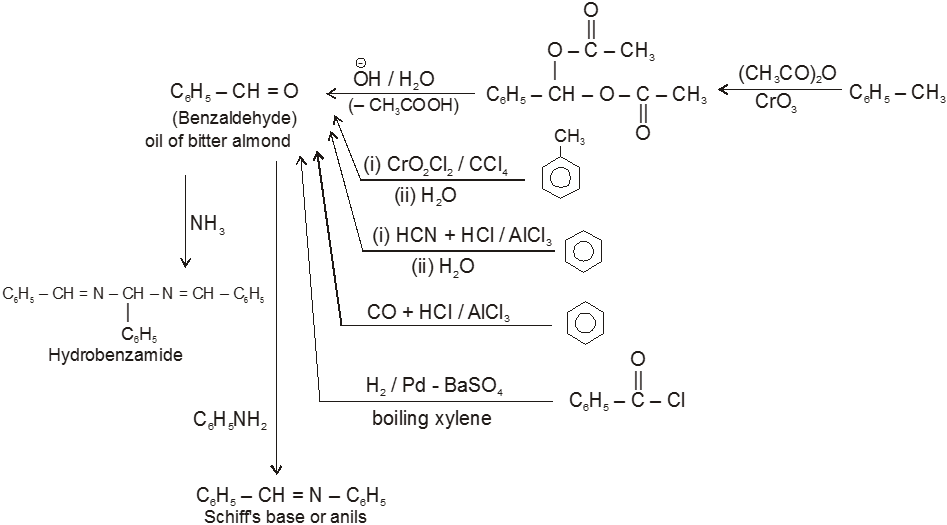
(V) 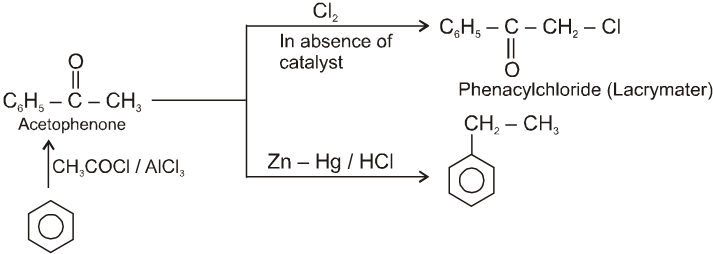
7. Uses of Aldehydes and Ketones
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Uses of Aldehydes and Ketones
- Act as solvents.
- Act as starting materials and reagents for the synthesis of other products.
- Formalin (40% solution of formaldehyde)- Used for preserving biological specimens manufacturing of bakelite, urea , formaldehyde glues and other polymers products.
- Acetaldehyde used in the manufacture of acetic acid, ethyl acetate, vinyl acetate, polymers and drugs.
- Benzaldehyde used in perfumery and in dye industries.
- Butyraldehyde, vanillin, camphor, etc., are well known for their odours and flavours.
- Acetone and ethyl methyl ketone are common industrial solvents.
8. Carboxylic acid and derivatives of carboxylic acids
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Carboxylic acid and derivatives
Introduction :
Compounds containing the carboxyl group are distinctly acidic and are called carboxylic acids.There general formula is CnH2nO2.
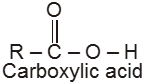
Carboxylic acid derivatives are compounds with functional groups that can be converted to carboxylic acids by a simple acidic or basic hydrolysis. The most important acid derivatives are esters, amides, nitriles, acid halides and anhydrides.
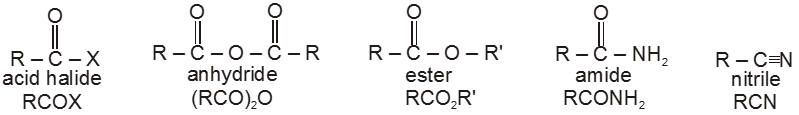
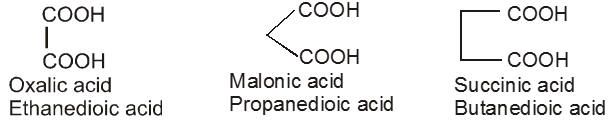
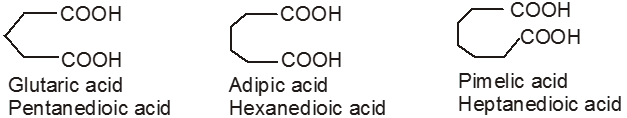
Dicarboxylic acids :
If the compound containing two carboxyl groups, these are known as dicarboxylic acid.
For example :
Physical properties of acids and acid derivatives :
(1) Boiling point : The boiling point of carboxylic acids are higher than that of alcohols, ketones or aldehydes of similar molecular weight.
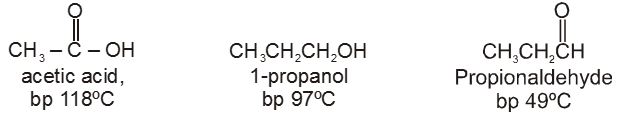
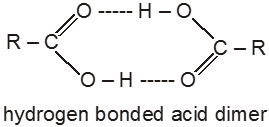
The high boiling points of carboxylic acids is the result of formation of a stable hydrogen-bonded dimer.
(2) Melting points :
Melting point of carboxylic acids : There is no regular pattern in melting point of carboxylic acid (up to 10 carbon atoms) having even number of C atoms are higher than neighbouring members having odd number of C atoms because carboxylic acid and methyl group in even members lie in opposite side of zig-zag carbon chain hence they fit better into crystal lattice resulting in higher melting points.Vice-versa is observed in case of carboxylic acid having odd no. of carbon atoms.

Amides have surprisingly high boiling points and melting points compared with other compounds of similar molecular weight. Primary and secondary amides participate in strong hydrogen bonding.
(3) Solubility:
Carboxylic acids form hydrogen bonds with water and the lower molecular - weight carboxylic acids (upto 4 carbon atoms) are miscible with water.
Acid derivatives (esters, acid chlorides, anhydrides, nitriles and amides) are soluble in common organic solvents such as alcohols, ethers, chlorinated alkanes and aromatic hydrocarbons. Acid chlorides and anhydrides cannot be used in nucleophilic solvents such as H2O and alcohols, because they react with these solvents.
(4) Aliphatic carboxylic acids upto nine carbon atoms are colourless liquids at room temperature, higher acids are wax like solids due to their low volatility.
9. Methods of Preparation of Carboxylic Acid
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Methods of Preparation of Carboxylic Acid
1. From primary alcohols
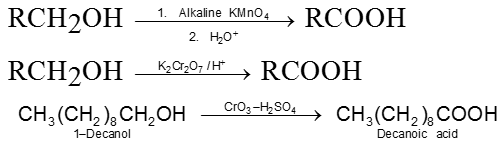
2. From aldehydes
![]()
Oxidising agents - HNO3, KMnO4, K2Cr2O7
Mild oxidising agents - Tollen’s reagent and Fehling’s reagent
3. From alkyl benzenes
![]()
1° and 2° alkyl benzene are oxidised in this manner. Tertiary group is not affected.
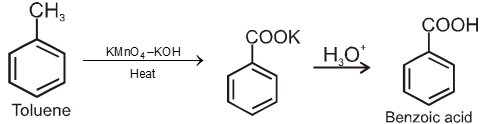
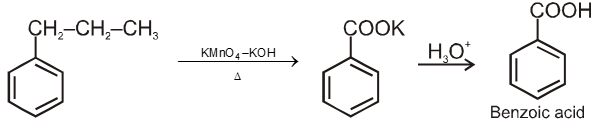
4. From nitriles and amides
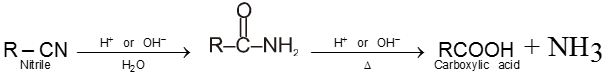
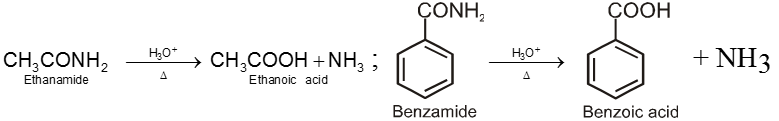
5. From Grignard reagents
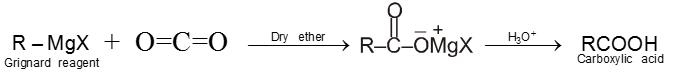
6. From acyl halides
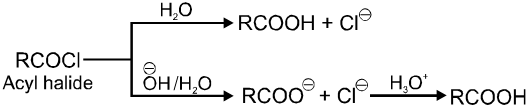
7. From acyl anhydrides
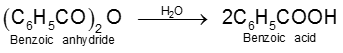
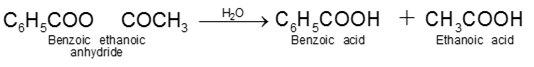
8. From esters
![]()
Ex.
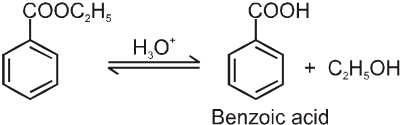
![]()
Ex. 
10. Chemical Reactions of carboxylic acid
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chemical Reactions of carboxylic acid
Acidic Nature : Acidity of carboxylic acids :
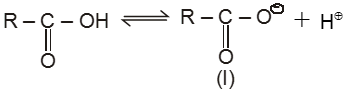
(i)![]()
(I) exists as two equivalent canonical structures I(A) and I(B). This ion is resonance stablised and resonance hybrid structure is I(C).
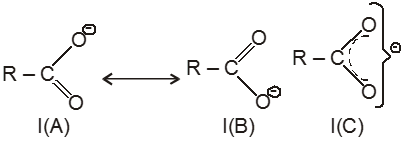
(ii) ion is more stable due to resonance, hence carboxylic acids are acidic in nature.
(iii) Electron withdrawing group (–I effect) stablises the anion and hence, increases acidic nature.
Ex. 
Ex. 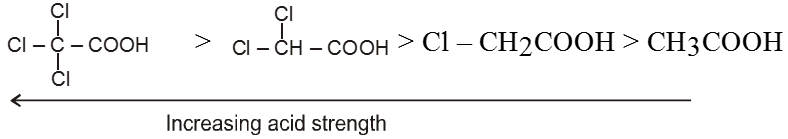
(iv) Electron releasing group (+ I effect) destablises the anion and hence decreases acidic nature.
Ex. HCOOH > CH3COOH > CH3 – CH2 – COOH
Ex. 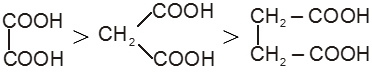
Direct attachement of groups such as phenyl or vinyl to the carboxylic acid increases its acidity.
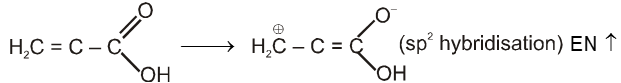
Ex. Relative acid strength is: RCOOH > HOH > ROH > HCCH > NH3 > RH
Note : Acidity of acids is compared by compairing stability of conjugate base.
(2) Reaction involving removal of proton from –OH group :
(i) Action with blue litmus : All carboxylic acids turn blue litmus into red.
(ii) Reaction with metals :
![]()
![]()
(iii) Reaction with alkalies :
CH3COOH + NaOH¾® CH3COONa + H2O
(iv) Reaction with carbonates and bicarbonates :
2CH3COOH + Na2CO3¾®2CH3COONa + CO2 + H2O
CH3COOH + NaHCO3¾®CH3COONa + CO2 + H2O
Reaction of carboxylic acid with aqueous sodium bicarbonate solution produces brisk effervescence. However most phenols do not produce effervescence. Therefore, the reaction may be used to distinguish between carboxylic acids and phenols.
(v) Reaction with grignard reagent :
![]()
Feasible reaction : A stronger acid displaces a weaker acid from the salt of the weaker acid.
Ex. CH3COOH (Stronger acid) + CH3ONa¾® CH3COONa + CH3–OH (Weaker Acid)
Ex. CH3COOH (stronger acid) + NaHCO3¾® CH3COONa + H2CO3 (Weaker acid) ¾® H2O + CO2![]()
(lab. test of carboxylic acid)
(3) Reaction involving replacement of –OH group :
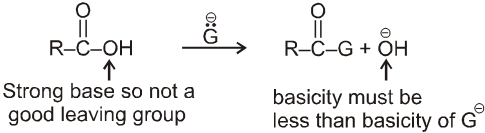
(i) Formation of acid chlorides :
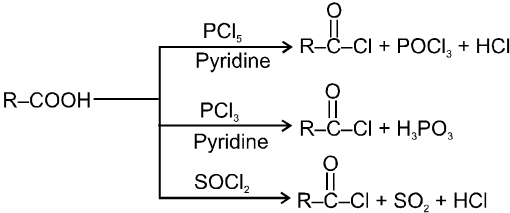
Ex.
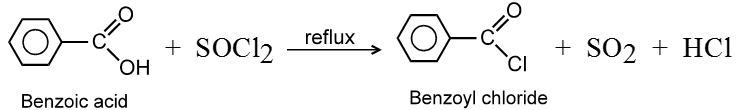
Ex.
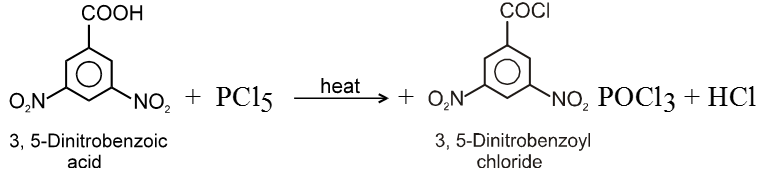
(ii) Fisher Esterification :
Carboxylic acid react with alcohol to form esters through a condensation reaction known as esterification.
General Reaction :
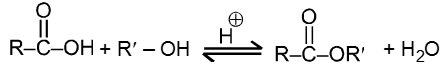
Specific Examples:
![]()
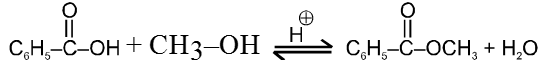
Mechanism : (Acid catalysed esterification)
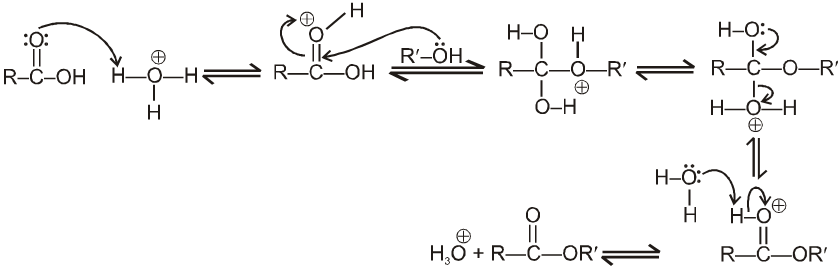
If we follow the forward reactions in this mechanism, we have the mechanism for the acid catalysed esterification of an acid. If however, we follow the reverse reactions, we have the mechanism for the acid catalysed hydrolysis of an ester.
Acid catalysed ester hydrolysis.
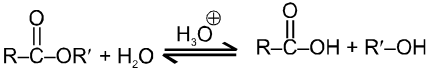
Which depends upon condition we choose. If we want to esterify an acid, use an excess amount of the alcohol and, if possible remove the water as it is formed.
If we want to hydrolyse an ester, we use a large amount of water i.e. dil HCl or dil H2SO4.
(iii) Formation of amides :
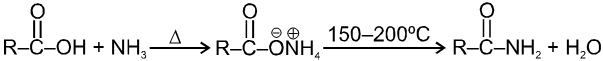
In fact amides can not be prepared from carboxylic acids and amines unless the ammonium salt is heated strongly to dehydrate it. This is not usually a good method of preparing amides.
(iv) Formation of acid anhydride :
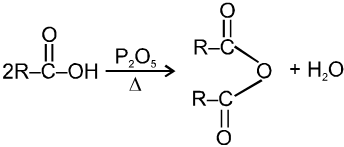
(4) Decarboxylation reactions :
(i)Soda-lime decarboxylation :
General reaction :
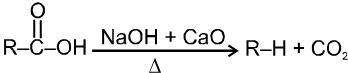
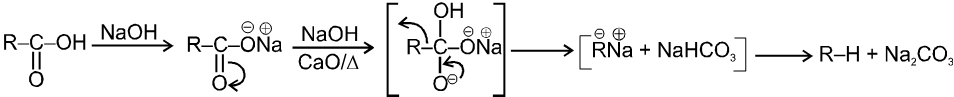
In this reaction carbanion intermediate is formed.
Rate of reaction depends upon the stability of carbanion intermediate.
Electron withdrawing group at R–COOH will increases the rate of decarboxylation.
Ex.
Rate of decarboxylation. I > II > III > IV > V
Ex.
Rate of decarboxylation. I > II > III > IV
(ii) Decarboxylation of b-keto carboxylic acids:
b-keto acids decarboxylate readily when they are heated to 100–150ºC.
![]()
There are two reasons for ease of decarboxylation.
When the acid itself decarboxylates, it can do so through a six membered cyclic transition state :
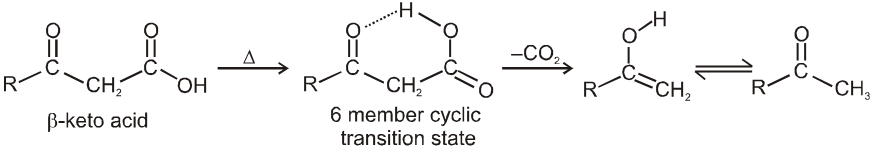
This reaction produces an enol directly and avoids an anionic intermediate. The enol then tautomerises to a methyl ketone.
When the carboxylate anion decarboxylates, it forms a resonance stabilized enolate anion.
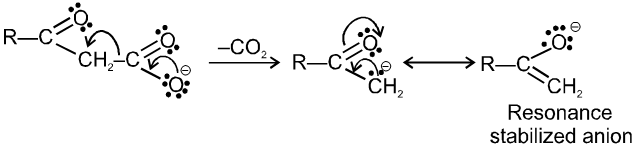
Alphatic acids that do undergo successful decarboxylation have certain functional groups or double or triple bonds in the a or b positions.
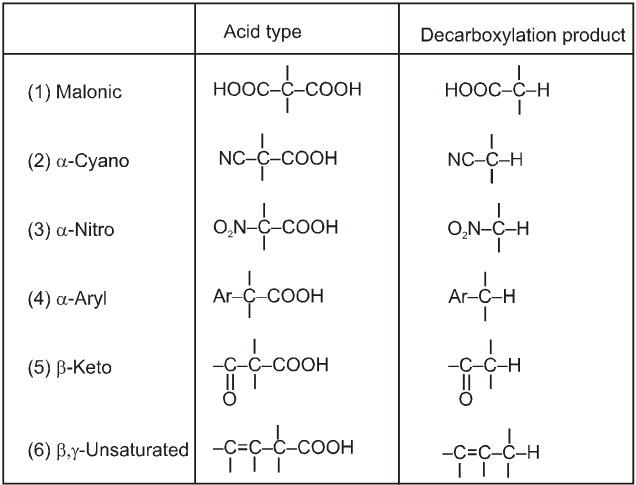
(iii) Kolbe’s electrolysis
![]()
If n is the number of carbon atoms in the salt of carboxylic acid, the alkane formed has 2(n–1) carbon atoms.
Ex.
![]()
(iv) Hunsdiecker Reaction (Bromo-decarboxylation) :
R–COOAg + Br2 ¾®R–Br + CO2 + AgBr
Heavy metal salt of acid is heated with Br2 it results in decarboxylations.
Metal can be silver ion, mercuric ion, or lead (IV) ion.
Mechanism :
Step 1 : ![]()
Step 2 : ![]()
Step3 : ![]()
Step 4 : ![]()
Although bromine is the most often used halogen, chlorine and iodine have also been used.
When iodine is the reagent, the ratio between the reactant is very important and determines the product.
A 1 : 1 ratio of salt to iodine gives alkyl halide, as above. A 2 : 1 ratio, however gives the ester RCOOR. This is called Simonini reaction and sometimes used to prepare carboxylic ester. The yield of alkyl halide follows the order Primary > Secondary > Tertiary
(5) HVZ Reaction (Halogenation of aliphatic acids and Substituted acids) :
In the presence of a small amount of phosphorus, aliphatic carboxylic acids react smoothly with chlorine or bromine to yield a compound in which -hydrogen has been replaced by halogen.
![]()
Summary of reactions of carboxylic acids :
11. Uses of Carboxylic Acids
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Uses of Carboxylic Acids
- Methanoic acid - In rubber, textile, dyeing, leather and electroplating industries.
- Ethanolic acid - As a solvent and as a vinegar in food industry.
- Hexanoic acid - In the manufacture of nylon-6, 6.
- Higher fatty acids - For the manufacture of soaps and detergents.
- Esters of benzoic acid - In perfumery.
- Sodium benzoate - As a food preservative.
2. Aniline
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
ANILINE (C6H5NH2)
General Methods of Preparation
(1) Lab method :
![]()
(2) Industrial method :
![]()
(3) From phenol :
![]()
(4) From benzamide (Hoffmann's bromamide reaction) :
C6H5CONH2 + Br2 + 4KOH ¾® C6H5NH2 + K2CO3 + 2KBr + 2H2O
(5) Form benzoic acid (Schmidt reaction) :
![]()
(Hydrazoic acid)
(6) From Grignard reagent :
![]()
(7) From phenyl isocyanide :
![]()
Physical properties
(i) Fresh, aniline is a colorless oily liquid. On standing the colour becomes dark brown due to action of air and light.
(ii) It's B.P. is 183ºC.
(iii) It is heavier than water.
(iv) It has characteristic unpleasent odour. It is toxic in nature.
Reactions due to –NH2 group
(1) Basis nature : Aniline is weak base but it forms salt with strong acids. It accepts a proton.
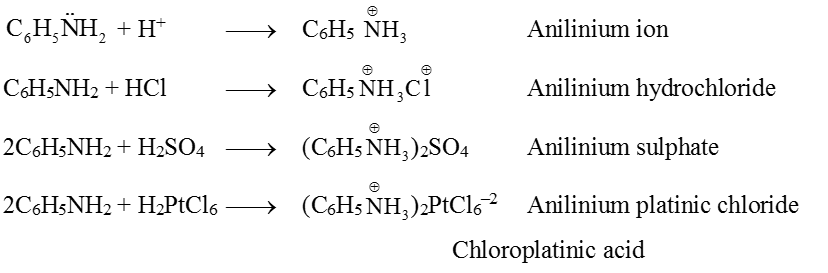
(2) Alkylation : Aniline reacts with alkyl halides forming secondary, tertiary and quaternary ammonium salts depending on the concentration of alkyl halides.
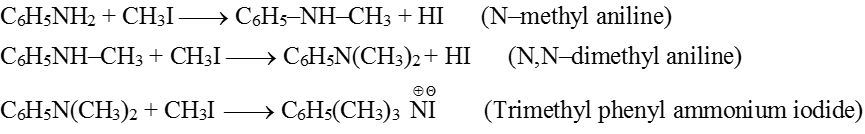
(3) Acylation : Aniline reacts with acid chlorides or anhydrides to form corresponding amides called anilides. [The reaction of C6H5NH2 with benzoyl chloride is example of "Schotten Baumann reaction"]
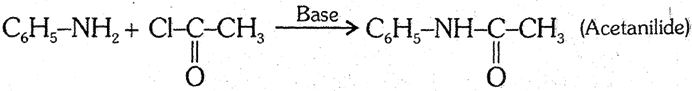
(4) Carbylamine reaction :
C6H5NH2 + CHCl3 + 3KOH ¾®C6H5NC + 3KCl + 3H2O
Phenyl isocyanide (Foul smell compound)
Note :
(1) Intermediate species is dichloro carbene [: CCl2].
(2) This is test of aniline and other primary amine, known as Isocynide test.
(5) Hoffmann's mustard oil reaction : When aniline is heated with alc. CS2 and excess of HgCl2 Phenyl isothiocyanate having a characteristic smell of mustard oil is formed.
This is a test of aniline and other primary amines.
(6) Reaction with aldehydes : Aniline condenses with aldehydes to form schiff's base.
(7) Reaction with hinsberg's reagent :
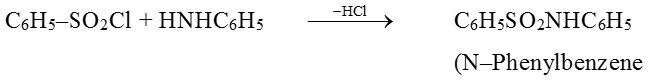
(8) Diazotisation : Diazotisation is a reaction in which ice cooled solution of aniline in an inorganic acid reacts with sodium solution leading to the formation of diazonium salt.
![]()
Benzene diazonium chloride is a useful synthetic reagent. It is used in the preparation of many organic compounds.

(1) Halogenation : In Polar and nonpolar medium Chlorine and bromine react with aniline and form trichloro and tribromo aniline respectively.

Note : However, monobromo or chloro derivative of aniline can be prepared if –NH2 group is first protected by acetyl group. Here the reactivity decreases due less +M effect on benzene ring.
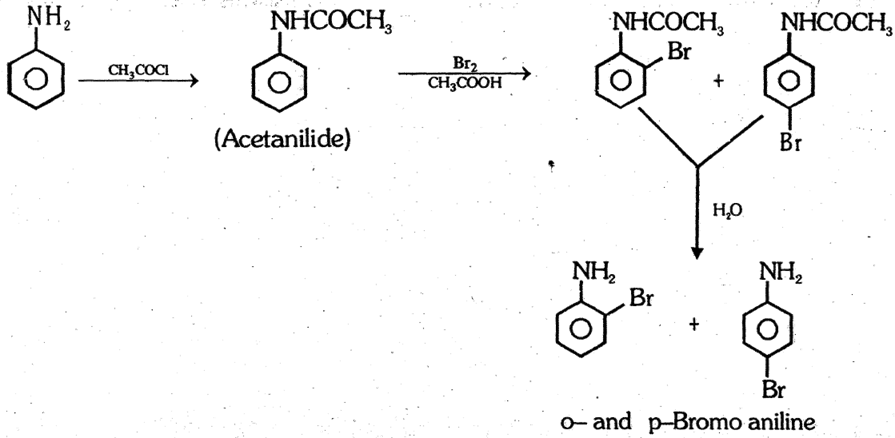
(2) Nitration :
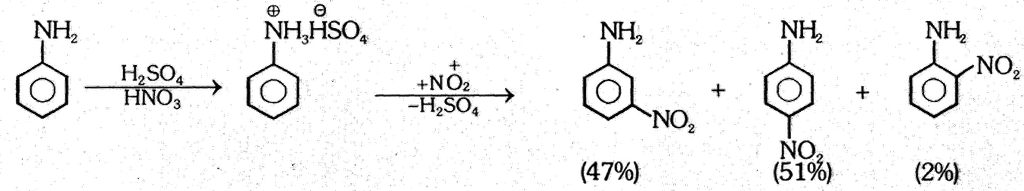
(3) Sulphonation : Aniline reacts with fuming H2SO4 to give sulphanilic acid. (p-Amino-benzene sulphonic acid)
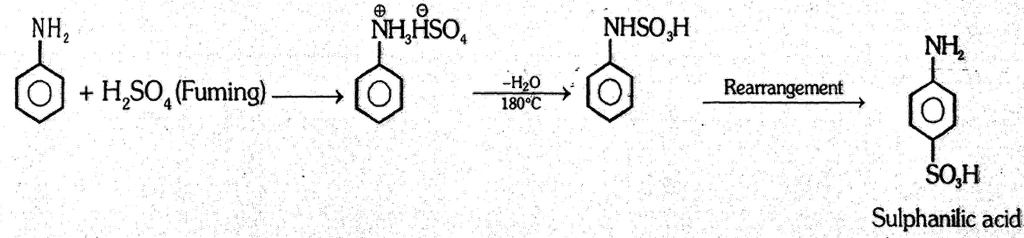
- This process is called baking.
- Sulphanilic acid is an important intermediate in the manufacturing of dyes and durgs.
- The compounds in which both proton donating & proton accepting groups present are called ampholite (dipolare ion)

(4) Catalytic hydrogenation: Aniline undergoes hydrogenation in presence of Ni at high temp. to form cyclohexanamine.

Tests of aniline
(i) Carbylamine test : Aniline gives carbylamine test or isocyanide test.
C6H5NH2 + CHCl3 + KOH ¾® C6H5NC (Bad smelling)
(ii) Dye test : Aniline is first diazotised. On adding alkaline solution. Of b-naphthol to the diaztised product a red-orange dye is formed.
(iii) On heating with bromine water, a white ppt. is formed.
2. Proteins
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Proteins
Proteins : Proteins are the most abundant-biomolecules of the living beings. The chief source of proteins are milk, cheese, pulses peanuts, fish meat etc. These are high molecular mass complex, biopolymers of amino acids. The word protein is derived from Greek word proteios which means primary or of prime importance.
Amino Acids : Each living cell is made up of thousands of different proteins All proteins contain the elements like carbon, hydrogen, oxygen, nitrogen and sulphur. Some of these also contain phosphorus, iodine and traces of metals such as, Fe, Cu, Zn, & Mn. All proteins are polymers of a-amino acids and on partial hydrolysis give peptides of varying molecular masses which upon complete hydrolysis give a-amino acids.
![]()
Type of a-amino acids :
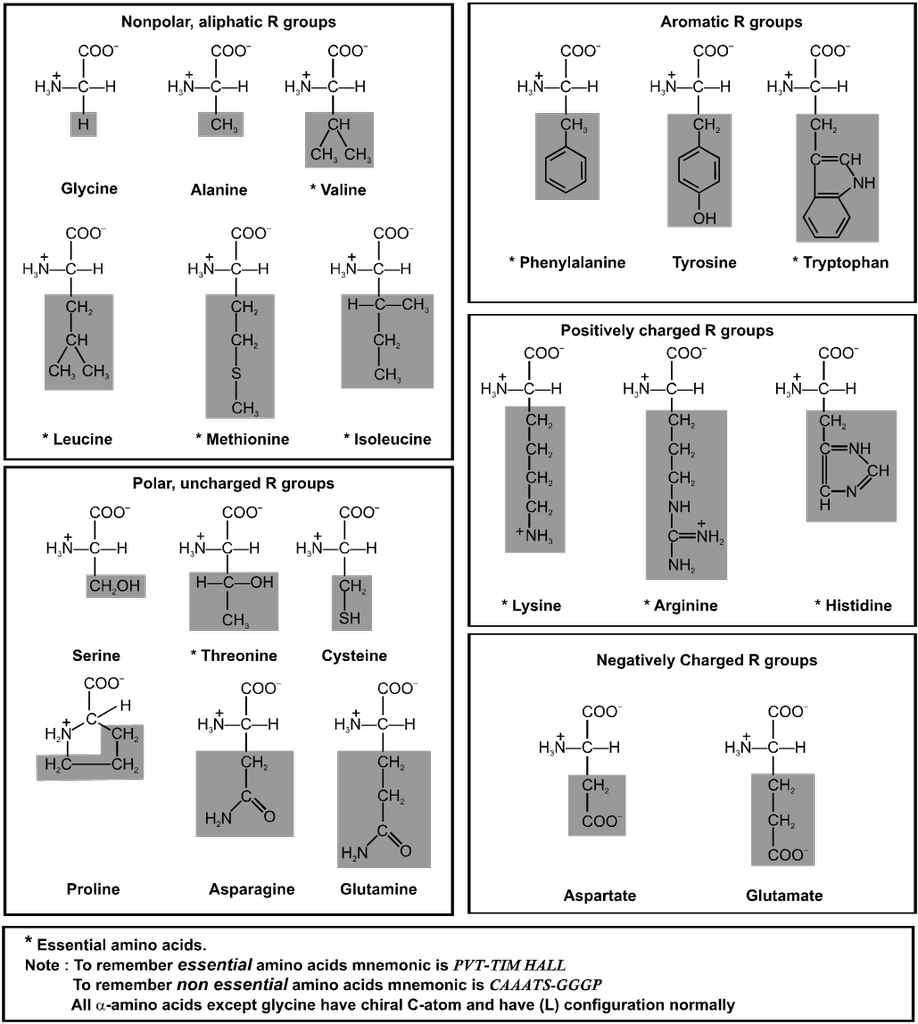
These organic compounds contain amino as well as carboxylic acid group. On the basis of position of amino group in the chain, these are named as a, b, g etc. amino acid.
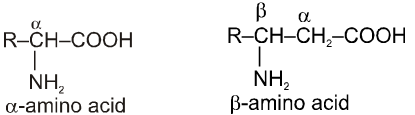
Amino Acid : Building Blocks of Proteins
Amino acids are the building blocks of the molecular structure of the important and very complex class of compounds known as proteins. The proteins on hydrolysis yield mixture of the component amino acids.
Amino acids are bifunctional compounds containing both an amino and a carboxylic acid group. They are represented by the general formula :
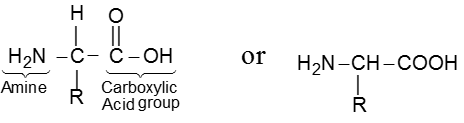
where, R = alkyl, aryl, or any other group.
Zwitter ion (Dipolar Nature of Amino acids) :
In a neutral amino acid solution, the –COOH loses a proton and the –of the same molecule picks up one.The resulting ion is dipolar, charged but overall electrically neutral. This is called Zwitterion (German, “two ions”). Therefore amino acids are amphoteric.
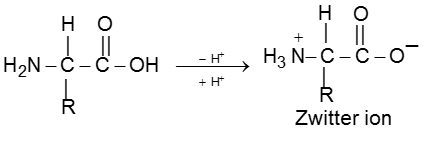
There are around 20 amino acids in the living system.
Classification of Amino Acids :
(A) On the basis of synthesis :
(i) Non essential amino acids : 10 amino acids are synthesis in our body and these are said to be non essential amino acids (eg. Gly, Ala, Glu, Asp, Pro and Cys).
(ii) Essential amino acid : 10 amino acids which are necessarily be present in our diet are called essential amino acids (eg. Val, Leu, I leu, Lys and Phe).
(B) On the basis of functional groups present :
(i) Neutral amino acids : If only one – NH2 and one – COOH groups are present. eg. Glycine, Alanine, Valine, Leucine etc.
(ii) Acidic amino acid : If one basic and two acidic groups are present. Additional acidic functional group must be present in the side chain. E.g. Aspartic acid and Glutamic acid.
(iii) Base amino acid : If two basic and one acidic group is present. Additional basic group must be present in the side chain. E.g. Arginine, Lysine & Histidine.
Isoelectric point of a-amino acids :
When an ionised amino acid is placed in an electric field, it will actually migrate towards the opposite electrode. Depending upon the pH of the medium, three things can happen. The positive form (II) will migrate to the cathode, the neutral form (I) (Zwitter ion) will not migrate, while the negative form (III) will migrate to the anode. The pH at which the amino acid shows no tendency to migrate when placed in an electric field is known as its isoelectric point. This is characteristic of a given amino acid. Thus glycine has its isoelectric point at pH 6.1.
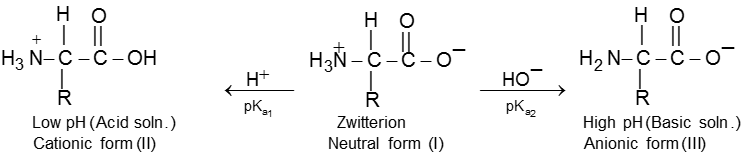
Isoelectric Point : The pH at which the amino acid shows no tendency to migrate when placed in an electric field is known as its isoelectric point.
Because of amphoteric nature in acidic solution it exist as the +ve ion. Hence it migrate towards cathode while in basic solution it exist as –ve ion and migrates towards anode.
In strongly acidic medium
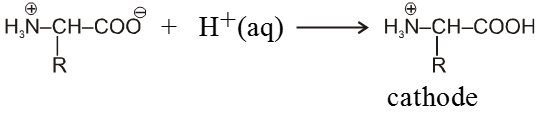
In strongly basic medium
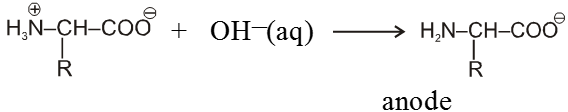
At some intermediate pH amino acids exist as a neutral dipolar ion i.e. the concentration of the cation and anions are equal and it does not migrate towards either electrode, this pH is called iso electric point of amino acid which is different for different amino acids.
For example :
(i) For neutral amino acid : pH of isoelectric point varies between 5.1 to 6.5. e.g. Glycine has pH value 6.0.
PI for neutral amino acid is calculated as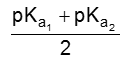
(ii) For acidic amino acid : Where there are two COOH groups and one NH2 group then isoelectric pH is around 3. e.g. Aspartic acid.
Aspartic acid :
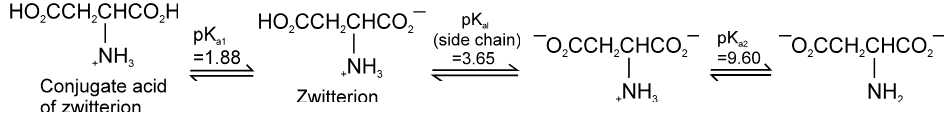
PI for acidic amino acid is calculated as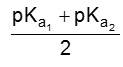
The pI of aspartic acid is the average of pKa1 (1.88) and the pKa of the side chain (3.65) or 2.77.
(iii) For basic amino acid : where there are two NH2 groups and one COOH group then isoelectric point varies between 7.6 to 10.8.
e.g. Lysine (9.8)
Lysine :
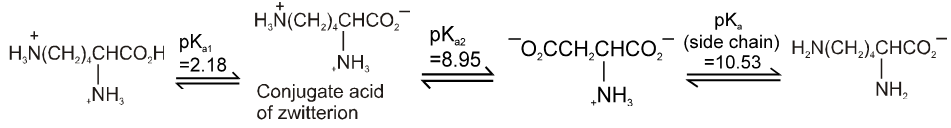
PI for basic amino acid is calculated as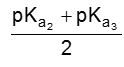
The PI of lysine is the average of pKa2 (8.95) and the pKa of the side chain (10.53) or 9.74.
Note : Amino acid has minimum aqueous solubilities at their isoelectric points.
(a) General methods of preparation
1. Aminolysis of a-halocarboxylic acid
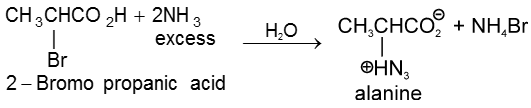
2. By strecker synthesis : Aldehyde reacts with a mixture of NH4Cl and NaCN to form a-aminonitrile (as an intermediate) which on hydrolysis gives an amino carboxylic acid.
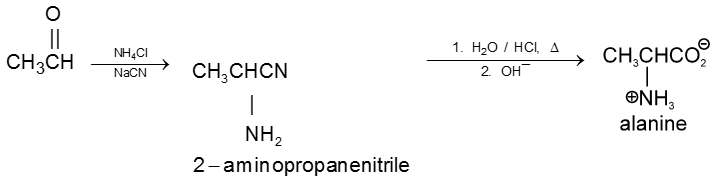
3. From aceto acetic ester :
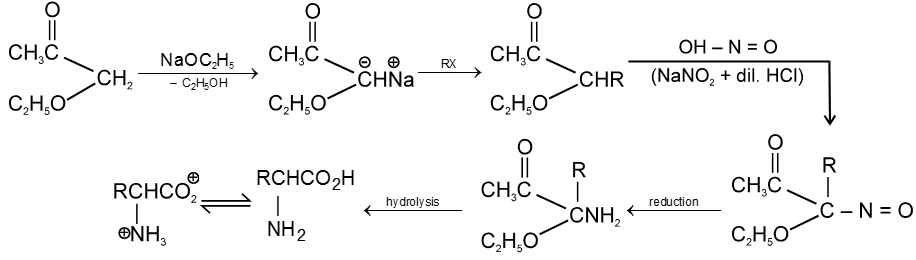
4. By Gabriel Synthesis :

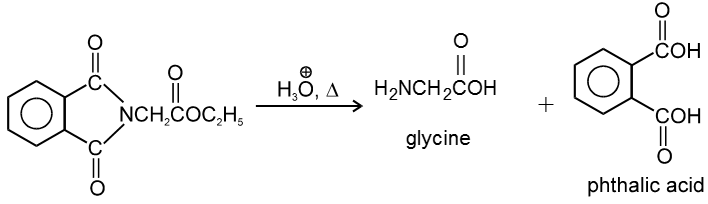
5. Effect of Heat :
a-amino acids undergo intermolecular dehydration on heating at about 200°C to give diketopiperazines.
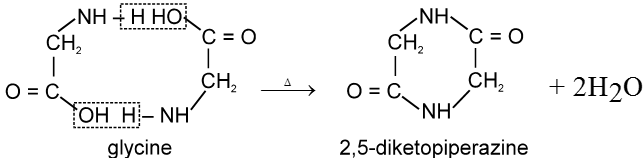
b-amino acids undergo intramolecular deamination on heating to form a, b-unsaturated acids.
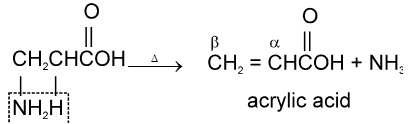
g-amino acids and d-amino acid undergo intramolecular dehyderation to form cyclic amides called.
Lactams.
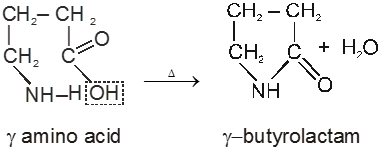
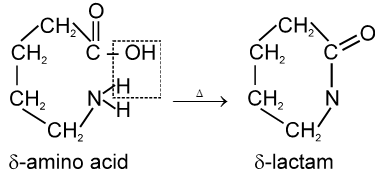
In case of d-amino acid, intramolecular cyclisation would given a seven-membered ring, which is formed with difficulty. Hence, there is intermolecular polymerisation forming nylon-6.
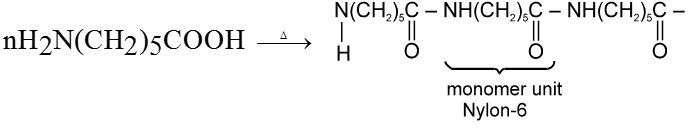
(b) Other Reactions of a-Amino Acid :
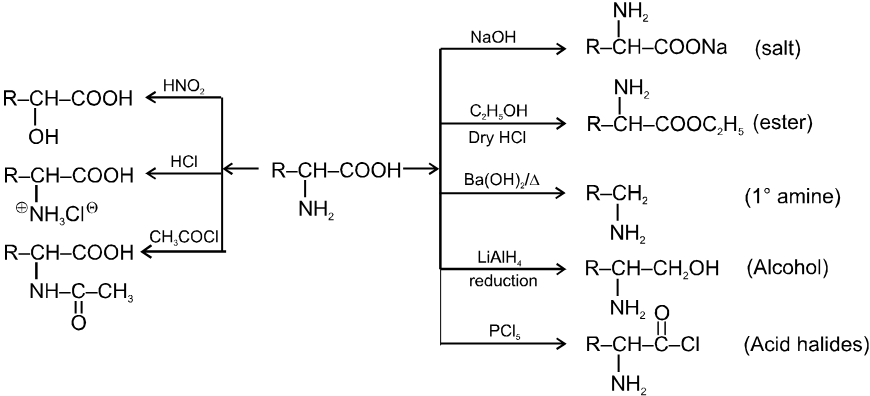
Peptide linkage :
Amino acid units are linked together by peptide ![]() – linkage and form polypeptides.
– linkage and form polypeptides.
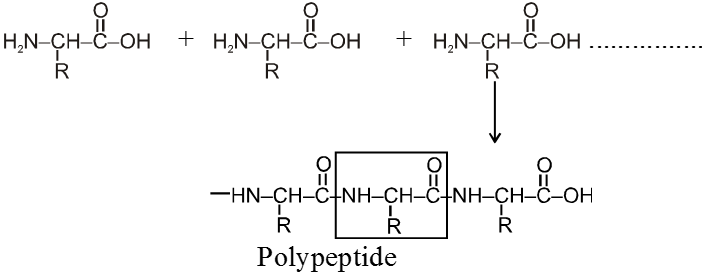
Note : Formation of protein linking together from different amino acids is not a random process. Each molecule of a given protein have the same sequence of amino acid along with its polypeptide chain. This tells about its own specific properties. Even the change of just one amino acid can change drastically the properties of entire protein molecules.
e.g. hemoglobin has 574 amino acids in definite sequence. The change of only one amino acid makes it defective hemoglobin which looses its property of carrying oxygen in blood stream and result a disease called sickel cell animea.
Writing and Naming of Polypeptides :
The structure of polypeptides are written in such a way that amino acid with free –NH2 group is written on the left hand side (N-terminal) of the polypeptide chain, while the amino acid with the free carboxyl group is written on the right hand side (C-terminal) of the chain. Thus a tripeptide alanylglycylphenylalanine (Al, Gly, Phe) is represented as follows :
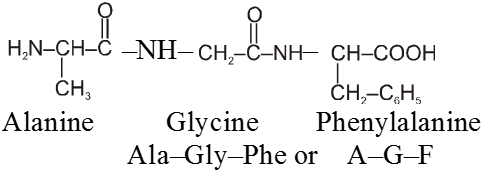
The name of any polypeptide is written starting from the N–terminal residue while writing the name the suffix “ine” in the name of the amino acid is replaced by “yl” (eg. glycyl for glycine, alanyl for alanine etc) for all the constituent amino acid except the C– terminal residue. This nomenclature is usually not used. Instead the three letter abbreviation or one letter codes for various a-amino acids present in the chain is used. For example the above tripeptide is named as Ala-Gly-Phe or A-G-F
Properties of polypeptides :-
(i) Polypeptides are amphoteric in character because of the presence of terminal ammonium and carboxylate ion as well as the ionised side chain of amino acid residues. Therefore like amino acids they neutralize both acids and bases and possess isoelectric points. At isoelectric point polypeptides have least solubility and hence can be separated.
(ii) The total contents of smaller peptides in tissues is small as compared to protein. However, like protein they also carry out many important functions in bio-system. Most of the toxins (poisonous substances) in animal and in plant sources are polypeptides. Some oligopeptides such as oxytoxin, vassopressin and angiotens are effective hormones while others act as sweetening agents. For example the dipeptide aspartyl phenyl alanine methyl ester called aspartame is 100-200 times more sweet than sucrose and is used as a suger substitute for dibetic patients
 Aspartame
Aspartame
Proteins :
Proteins are the most abundant-biomolecules of the living beings. The chief source of proteins are milk, cheese, pulses peanuts, fish meat etc. These are high molecular mass complex, biopolymers of amino acids. They occur in every part of the body and form fundamental basis of structures and determine functions of life. The word protein is derived from Greek word proteios which means primary or of prime importance.
Proteins are present in almost all the living cells of plant and animals. The protoplasm of plant and animal cells contains (10-20%) proteins. In human beings they are the main constituent of muscles, skin, hair, nail, tendons, arteries and connective tissue.
Classification of Proteins :
(I) On the basis of molecular structure :
(i) Fibrous proteins
(ii) Globular proteins
(i) Fibrous Proteins : When polypeptide chain run parallel and are held together by hydrogen and disulphide bonds then fiber like structure is formed such proteins are generally insoluble in water.
Ex. Keratin, myosine, collagen, etc.
(ii) Globular proteins : The polypeplide chain in these proteins is folded around it self to form spheroidal shape. The folding takes place in such a manner that lipophilic (non-polar, fat soluble) parts are pushed inwards and hydrophilic part (polar, water soluble) are pushed outwards. As a result water molecules interact strongly with the polar groups and hence globular proteins are soluble in water. As compare to fibrous proteins these are very sensitive to small changes in temperature and pH.
e.g. Insulin, albumin, thyoglobulin, antiadiodies, haemoglobin fibrinogen etc.
(II) On the basis of chemical composition :
(i) Simple proteins : These proteins on hydrolysis give only a-aminoacids. e.g. albumin in the white portion of eggs, glutenin in wheat, oxygenin in rice, keratin in hair, nails, horns etc.
(ii) Conjugated Proteins : These are the proteins in which protein part is combined with non-protein part. These proteins on hydrolysis give a non-protein in addition to the a-amino acids (Prosthetic group). The main function of prosthetic group is to control the biological function of the protein. Prosthetic groups are carbohydrate, phosphate group, lipids (ester of higher fatty acids) and so on. Casein of milk, haemoglobin of blood are conjugated proteins.
(iii) Derived Proteins : These are the degradation products obtained by partial hydrolysis of simple or conjugated proteins with acids, alkalies or enzymes. e.g. proteoses peptones, and polypeptides
Protein ® Proteoses ® Peptones ® Polypeptides
Structure of Proteins :
Proteins are biopolymers having a large number of amino acids through peptide bonds having three dimensional (3D) structures. Structure and shape of proteins can be studied and evaluated at four different levels i.e. primary, secondary, tertiary and quarternary, each level being more complex than the previous one.
(i) Primary structure of Proteins :
The manner and specific sequence in which different amino acids are joined to form polypeptides in a protein molecule is called 10 structure of that protein. Any change in this primary structure i.e sequence of amino acids creates a different proteins.
The first primary structure of a protein i.e INSULIN was determined by Frederic Sanger (a British chemist) and for this work he was awarded the Nobel prize in 1958. The different chemical and biological properties of various proteins are primarily due to the difference in their primary structures.
Normal Haemoglobin –Val – His – Leu – Thr – Pro – Glu –Lys–
Sickle cell anemia Val – His – Leu – Thr – Pro – Val – Lys–
(ii) Secondary structure of Proteins :
The conformation in which the polypeptides chains assume a shape as a result of H-bonding is called the secondary structure of protein.
Types:
(a) a-Helix structure
(b) b-pleated sheet structure
(iii)Tertiary structure of proteins :
The tertiary structure of protein represent overall folding of the polypeptide chains i.e further folding of the secondary structure. In other words, tertiary structure refers to the manner in which the entire protein molecule folds up in the three dimensional space to produce a specific shape. It gives rise to two major molecular shapes fibrous and globular. The main forces which stabilise the 2º and 3º structures of proteins are hydrogen bonds, disulphide linkages, vander waals and electrostatic forces of attraction.
(iv) Quarternary structure of proteins :
Although many proteins exist as a single polypeptide chain but there are certain proteins which are composed of two or more polypeptide chains referred to as sub-units or protomoss. These subunits may be identical or different and are held together by non-polar covalent forces such as, H-bonds, vanderwaals interaction and electrostatic interactions. The quarternary structure refers to the determination of the number of sub-units and their special arrangement w.r.t. each other in an aggregate protein molecule The Haemoglobin possesses quarternary structure and it is an aggregate of four polypeptide chains or sub-units.
A diagramatic representation of all four structure is given below, each ball represent an amino acid.
Denaturation of proteins :
A protein found in a biological sytem having a unique three dimensional structure and specific biological activity is called a native protein. When a protein in its native form is subjected to a physical change like change in temperature or pH, the hydrogen bonds are disturbed. As a result-globules unfold and helices get uncoiled and protein loses its biological activity. This is called Denaturation of proteins. During denaturation 2º and 3º structure are destroyed but 1º structure remain intact.
- Changing the pH denatures proteins because it changes the charges on many of the side chains. This disrupts electrostatic attractions and hydrogen bonds.
- Certain reagents such as urea and guanidine hydrochloride denature proteins by forming hydrogen bonds to the protein groups that are stronger than the hydrogen bonds formed between the groups.
- Detergents such as sodium dodecyl sulphate denature proteins by associating with the non polar groups of the protein, thus, interfering with the normal hydrophobic interactions.
- Organic solvents denature proteins by disrupting hydrophobic interactions.
- Proteins can also be denatured by heat or by agitation. Both increase molecular motion, which can disrupt the attractive forces. The coagulation of egg white on boiling and curdling of milk caused by the bacteria present in milk are common examples of denaturation of protein.
Renaturation of proteins :
The denaturation may be reversible or irreversible. The coagulation of egg white on boiling and curdling of milk are an example of irreversible protein denaturation. There is some cases where the process in actually reversible. The reverse process is called renaturation. In such cases when the temperature and pH of a denatured protein are brought back, the secondary and tertiary structures of protein are restored.
Test for proteins:
(a) Biuret test : Biuret reagent is a blue colour mixture of sodium hydroxide and copper sulphate. The blue colour of the reagent turns violet in the presence of proteins.
Condition :
- At least two peptide linkages must be present.
eg. Dipeptides do not give this test but tri, tetra,..........can give this test.
(b) Xanthoprotic:
Protein + conc. HNO3 ® Orange colour alkaline solution
(c) Ninhydrin test :
Proteins or Amino acids Purple coloured product.
3. Polymers
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Polymers
These are very high molecular mass substances where each molecule is derived from very large number of simple molecules joined together in a regular way. This simple molecule is monomer and the process of formation of polymers from simple molecule is polymerization.
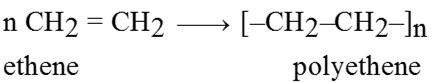
Polymers are two types :
(i) Homopolymers :
Polymers in which repeating structural units are derived from only one type of monomer units are called homopolymers.

Ex. Polypropylene, polyvinyl chloride (PVC), polyisoprene, neoprene (polychloroprene) polyacrylonitrile (PAN), nylon-6, polybutadiene, teflon (polytetrafluoroethylene), cellulose, starch etc.
(ii) Copolymers :
Polymers in which repeating structural units are derived from two or more types of monomer units are called copolymers.

Note : In Homo polymer empirical formula of monomer and polymers are same
4. Classification of Polymers
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Classification- Polymers can be classified by following ways
(A) Classification based on origin or source :
(B) Classification based on synthesis :

(C) Classification based on Structure:
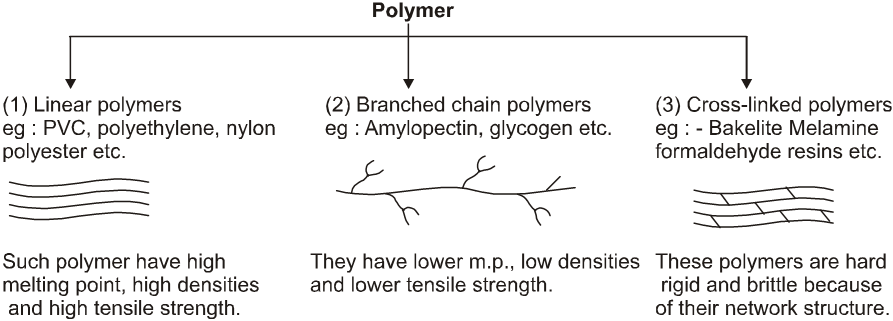
(D) Classification based on Mechanism :
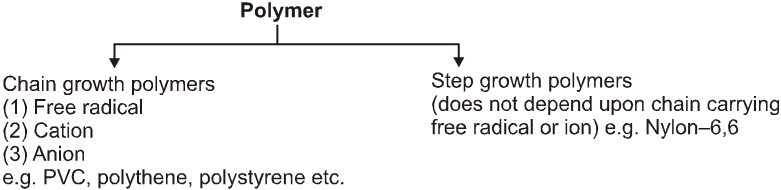
(1) Free radical :
This mechanism involve catalyst which generate free radical. Steps involved are :

(E) Calssification based upon molecular force :
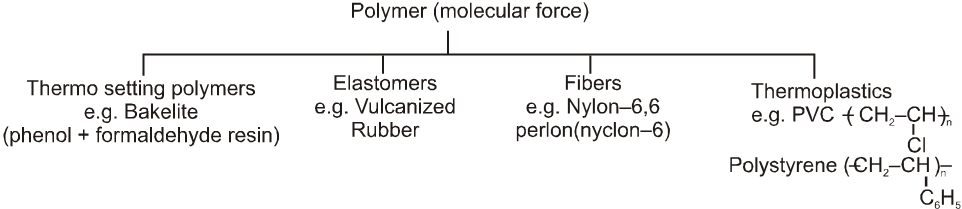
(i) Elastomers : Polymers in which the intermolecular forces of attraction between the polymer chains are the weakest are called elastomers.
e.g. Buna rubber, Natural rubber etc.
(ii) Fibres : Polymers in which the intermolecular forces of attraction are the strongest are called fibers. These forces are either due to H-bonding or dipole-dipole interactions. In case of nylon (polyamides), the intermolecular forces are due to H-bonding while in polyesters (terylene, dacron) and polyacrylonitrile (orlon, acrylin) Dipole-dipole interactions between the polar carbonyl (C = O) groups and, between carbonyl and cyano (– C º N) groups respectively.
(iii) Thermoplastics : Polymers in which the intermolecular forces of attraction are in between those of elastomers and fibres are called thermoplastics. The process of heat softening and cooling can be repeated as many times as desired without any change in chemical composition and mechanical properties of the plastic.
e.g. Polyethylene, polypropylene, polystyrene, PVC, teflon etc.
(iv) Thermosetting polymers : These are semifluid substances with low molecular weights which when heated in a mould, undergo change in chemical composition to give a hard, infusible and insoluble mass. This hardening on heating is due to extensive cross-linking between different polymer chains to give a three-dimensional network solid. Examples : Phenol-formaldehyde (bakelite), urea-formaldehyde, etc.
e.g. Bakelite urea-formaldehyde resin, terylene etc.

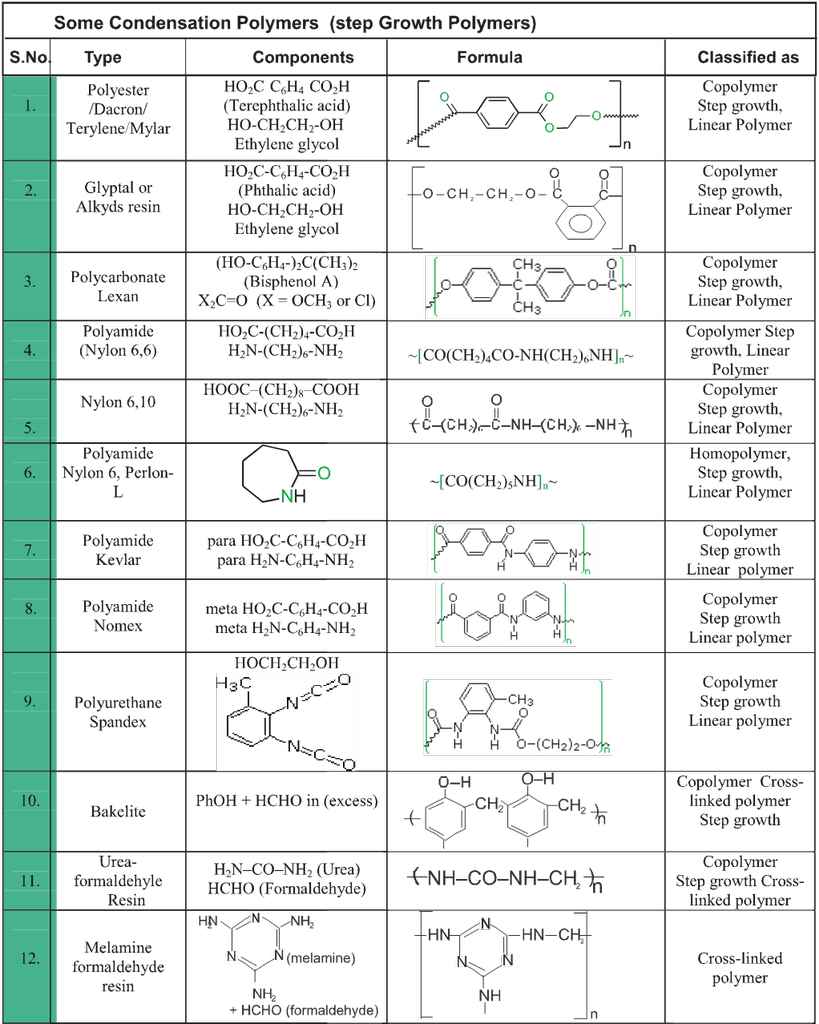

Natural rubber :
Natural rubber is a polymer of isoprene, and obtained from natural source-latex tree. In natural rubber, isoprene units are joined together in head-to-tail fashion and all double bonds in the polymer chain have cis configurations as shown in the given figure.
(Natural rubber)
The polymer contains cis repeating units and has a molecular weight ranging from 100,000 upto 1,000,000. A related polymer, called gutta percha which has a structure with trans double bonds and a much lower molecular weight. A typical sample of gutta percha has a molecular weight of about 7,000.
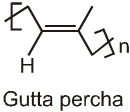
The cis arrangement of the double bonds in natural rubber prevents the rubber molecules from fitting into an ordered structure. Thus, rubber is an amorphous polymer. Because of the random coiling of its polymer chains, rubber stretches easily. When strectched, the rubber molecules are forced into a higher energy state. When the tension is released, rubber snaps back to its original random coiled state.
Vulcanization of Natural Rubber
Raw rubber is soft and tacky, they have very low mechanical strength, affected by environmental factors such as light, temperature, and oxygen and are of no use for any commercial purpose. These factors make rubber unsuitable for a number of applications. To enhance the mechanical properties and stiffness of natural rubber in 1839, charles Goodyear devised a method of reacting rubber with sulphur to form a more durable material.
Valcanization is a process in which natural rubber is heated with sulphur, where it undergo crosslinking and connect the isolated rubber chain through a network making them strong and stiff. Vulcanization forms both the cyclic structure shown on the left and the more desirable cross-linked structure shown on the right in the following figure.
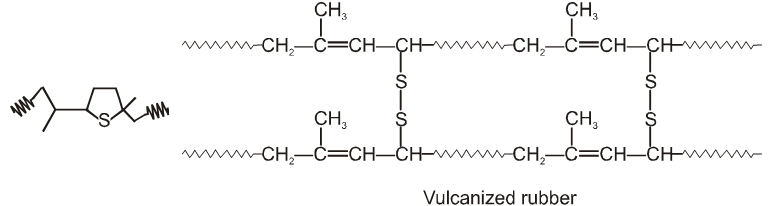
Increasing the amount of sulphur makes the vulcanized polymer harder and more durable. Adding 3-5% sulphur makes a product good for rubber bands and inner tubes. Adding 20-30% sulphur makes a hard rubber that was once widely used in ways that a hard synthetic plastic is used today. The vulcanization process made early automobile tires possible.
5. Biodegradable Polymers
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Biodegradable Polymers
A large number of polymers are quite resistant to the environmental degradation processes and are thus responsible for the accumulation of polymeric soild waste materials. These soild wastes cause acute environmental problems and remain undegraded for quite a long time. In view of the general awareness and concern for the problems created by the polymeric soild wastes, certain new biodegradable synthetic polymers have been designed and developed. These polymers contain functional groups similar to the functional groups present in biopolymers.
Aliphatic polyesters are one of the important classes of biodegradable poylmers. Some examples are given below :
(A) Poly b-hydroxybutyrate – co–b-hydroxy valerate (PHBV) :
It is obtained by the copolymerisatin of 3-hydroxybutanoic acid. PHBV is used in speciality packaging, orthopaedic devices and in controlled release of drugs. PHBV undergoes bacterial degradation in the environment.
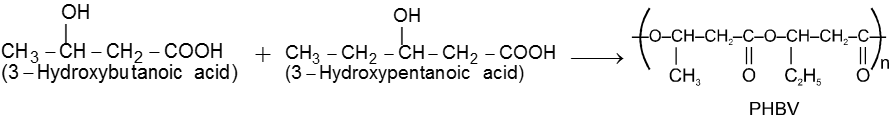
(B) Nylon–2–nylon–6 :
It is an alternating polyamide copolymer of glycine (H2N–CH2–COOH) and amino caproic acid
[H2N (CH2)5 COOH].
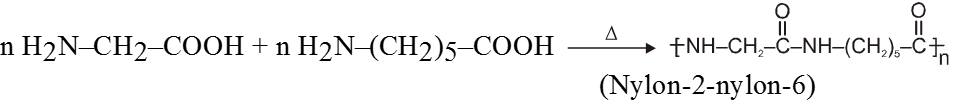
It is step-growth copolymer.
(C) Poly glycolic acid (PGA) and poly lactic acid (PLA) :- This copolymer is commercially called dextron.
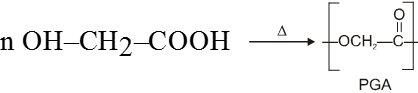
Glycolic acid
A copolymer of PGA and PLA (90 : 10) was the first biodegradable polyester used for stitching of wounds after operation.
(D) Poly-S-caprolactone lactone (PCL) : - It is obtained by chain polymerization of the lactone of 5(or S) hydroxy hexanoic acid.
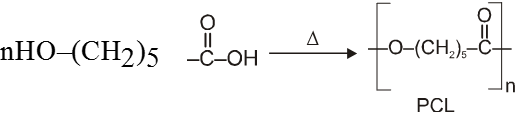
5(or S-) hydroxy hexanoic acid
2. Types of Chemical Messenger
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Types of chemical messengers
There are two types of chemical messengers :
- Hormones
- Neurotransmitters.
(i) Hormones :
Classification of hormones : Based on their chemical structure ; hormones fall into three classes.
Steroid hormones :
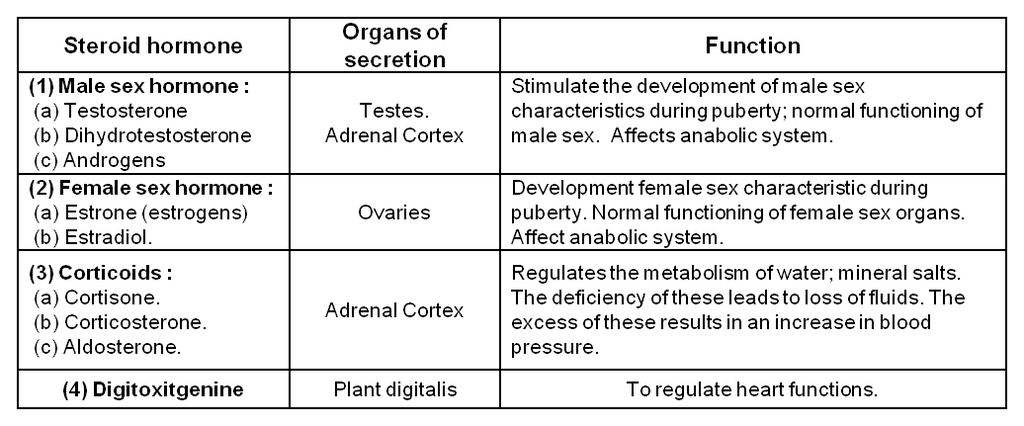
Peptide hormones :

Amine hormones :
Those which are neither steroids nor peptides. These are water soluble amine compounds.
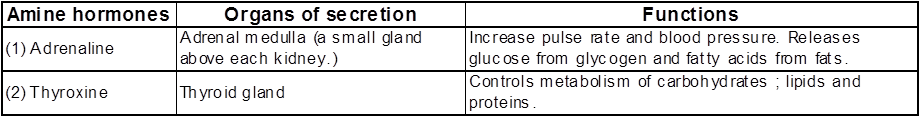
(ii) Neurotransmitters :
*Nerves transfer message through neurotransmitters.
*There are small molecules such as acetyl choline, dopamine and serotonin.
3. Drugs According to their Action
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Drugs According to their Action :
(1) Antacids :
*The chemicals which are used to reduce the acidity of the stomach are called antacids.
*Antacids are nature is basic. Their PH value is in the range of 7.0 to 8.0.
eg. : Omeprazole, Lansoprazole
(2) Antihistamines or Antiallergic Drugs :
*Antihistamines are the drugs which diminish or abolish the effects of histamine.
eg. : Histamine, Cimetidine, Ranitidine
Allergy :
Allergy may be defined as the hypersensitive reponse of the body of certain persons to the external stimulus (such as some drugs, foods, dust, pollen grains, catfur, fabrics etc.)
*The substances which cause allergy are called allergens.
*Most commonly used anti-histamine under the trade name avil :
(3) neurologically Active Drugs
(a) Tranquilizers :
*The chemicals which are used to reduce mental tension, relieve anxiety and mental stress are called Tranquilizer. They act on central nervous system and are hypnotics.
*Tranquilizers are effective in such mental disorder when ordinary hypnotics or sedatives fail. These are called as psychotherapeutic drugs.
eg. : Barbituric acid, Luminal, Serotonin, Bithional
Note : * Reserpine, an alkanoid, is a powerful tranquillzer. It is obtained from a plant, Rauwolfia serpentina (common name - Sarpagandha) which grows in india.
(b) Analgesics : The chemicals which are used for relieving pain are called ANALGESICS.
(i) Non-narcotic analgesics (Non addictive) : Aspirin and Paracetamol belong to this class.
*These drugs also act as antipyretic (reducing fever), and preventing platelet coagulation.
(ii) Narcotic Analgesics :
*Morphine and its homolgues in medicinal doses, relieve pain and produce sleep. In higher doses these produce STUPOR , COMA, CONVULSIONS and ultimately death.
*The narcotics are mainly used for the relief of postoperative pain, cardiac pain and pain of terminal cancer, and in child birth.
eg. : 2-Acetoxy benzoic acid (Aspirin), Paracetamol (4-acidamido phenol), Ibuprofen
(4) Antimicrobials :
*The chemicals which stop the growth or kill the micro organism such as bacteria, virus, fungi, moulds etc are called antimicrobials.
* Antibiotics, antiseptics and disinfectants are antimicrobial drugs.
(a) Antibiotics : *The chemicals produced by micro organisms like bacteria, fungi and moulds that inhibit the growth or destory other micro organism causing infectious diseases in men or animal’s body are called antibiotics. *Alexander fleming in 1929 first.
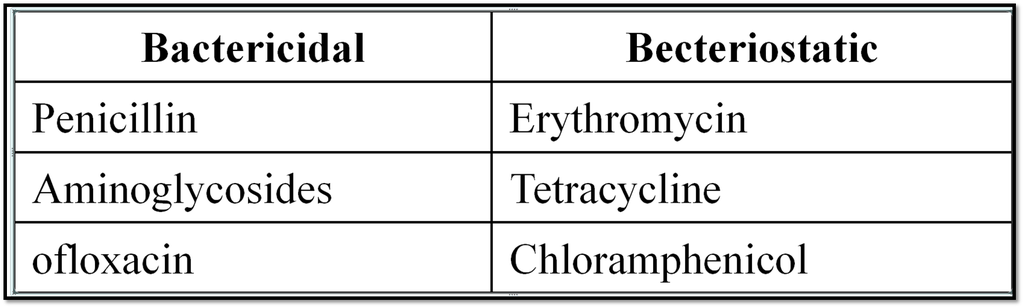
(I) Penicillin : *Six types of penicillines have been isolated so far. Among them penicillin-G is most widely used and is narrow spectrum.
*Ampicillin and amoxicilline are synthetic modification of penicilline and these has broad spectrum effect.
Penicillin is used for the treatment of pneumonia bronchitis bounds etc.
(II) Streptomycin : It is an effective broad spectrum antibioctic. It is used for the treatment of tuberculosis, meningitis and pneumonia
(III) Tetracyclins : Teramycin and oriomycin are important examples of this class of antibiotics. Teramycin is used for the treatment of typhoid and oriomycin is used for the the treatment of eyes.
(IV) Chloramphenicol : It is marketed as chloromycetin and is used for the treatement of typhoid, dysentery pneumonia, meningitis etc.
(b) Antiseptic and Disinfectants :
*Antiseptics are applied to the living tissues such as wounds, cuts ulcers and diseased skin surface. Examples are furacin, soframycin etc.
*Commonly used antiseptic is detol, it is a mixture of chloroxylenol and terpineol.
*Bithional is added to soaps to impart antiseptic properties.
*Iodoform is also used as antiseptic for wounds, boric acid in dilute aqueous solution is weak antiseptic for eyes.
Disinfactants are the substances which applied to inanimate objects such as floors drainage system instruments etc.
*One substance can act as an antiseptic and also act as disinfactant for example
(a) 0.2 percent solution of phenol is an antiseptic while its 1% solution is disinfectant.
(b) Chlorine in 0.2 to 0.4 ppm in aqueous solution is used to disinfect drinking water.
(c) Hexachlorophen :- It is mainly used in soaps creams and emulsions.
(d) Thymol : It is a natural derivative of phenol and is a powerfull disinfectant
(e) Amyl meta cresol (5-methyl-2-pentyl phenol) it is used as antiseptic in mouth wash or gargles.
(f) Gention violet and methylene blue are organic dyes but used as effective antiseptic.
(c) Sulpha Drugs :
A group of drugs which are derivatives of sulphanilamide are known as sulpha drugs.
eg. : Sulphadiazine, Sulphapyridine
(d) Antimalarials :
*These are the medicines of malaria. In earlier days malaria was treated with the bark of cinchona tree.
* The chloroquine phosphates are sold in the market under the trade name - resochin, ciplaquine, nivaquine etc.
(e) Antifungal Drugs :
*These are drugs used for superfical and deep (systemic) fungal infections.
*Two important antibiotics used as antifungal drugs, introduced way back in 1960, are amphotericin-B and griseofulvin.
(f) Antiamoebic Drugs :
*These are drugs useful in infection, caused by the protozoa Entamoeba histolytica.
*Metronidazole, tinidazole and tetracyclines are important antiamoebic drugs, used these days.
(g) Antiviral Drugs :
* Viruses are the ultimate expression of parasitism; they not only take nutrition from the host cell but also direct its metabolic machinery to synthesize new virus particles.
* Acyclovir, ribavirin, zidovudine, interferons are some of the important antiviral drugs, used these days.
(5) Antifertility Drugs :
“Chemcial substances which are used to check pregnancy in women are called anti-fertility drugs or birth control pills or oral contraceptives”.
*Birth control pills essentially contain a mixture of synthetic estrogen and progesteron derivatives. Both of these compounds are hormones.
eg. : Norethyndron, Ethynylestradiol (novestrol)
*Mifepristone is a synthetic steriod that blocks the effects of progesterone and is used as a “morning after pill” in many countries.
eg. : Mifepriston
4. Chemicals in Food
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chemicals In Food
Chemicals are added to food for their preservation, enhancing their appeal and adding nutritive values in them Main catergories of food additives are as follows
(i) Food colours.
(ii) Flavours and sweeteners.
(iii) Fat emulsifiers and stabilising agents.
(iv) Flour improvers antistaling agent and bleaches.
(v) Antioxidants
(vi) Preservatives
(vii) Nutritional supplements such as minerals, vitamins and amino acids except category
None of the above have nutritive values.
(a) Artificial sweetening agents :
* Saccharine is the first popular artificial sweetening agent used since 1879. It is about 550 times more sweet as cane sugar.
*It’s use is of great value to diabetic persons and people who need to control intake calories.
*It is used in pan masala cheap Ice cream, cheap drinks, mouthwash, cheap toffies, toothpaste).
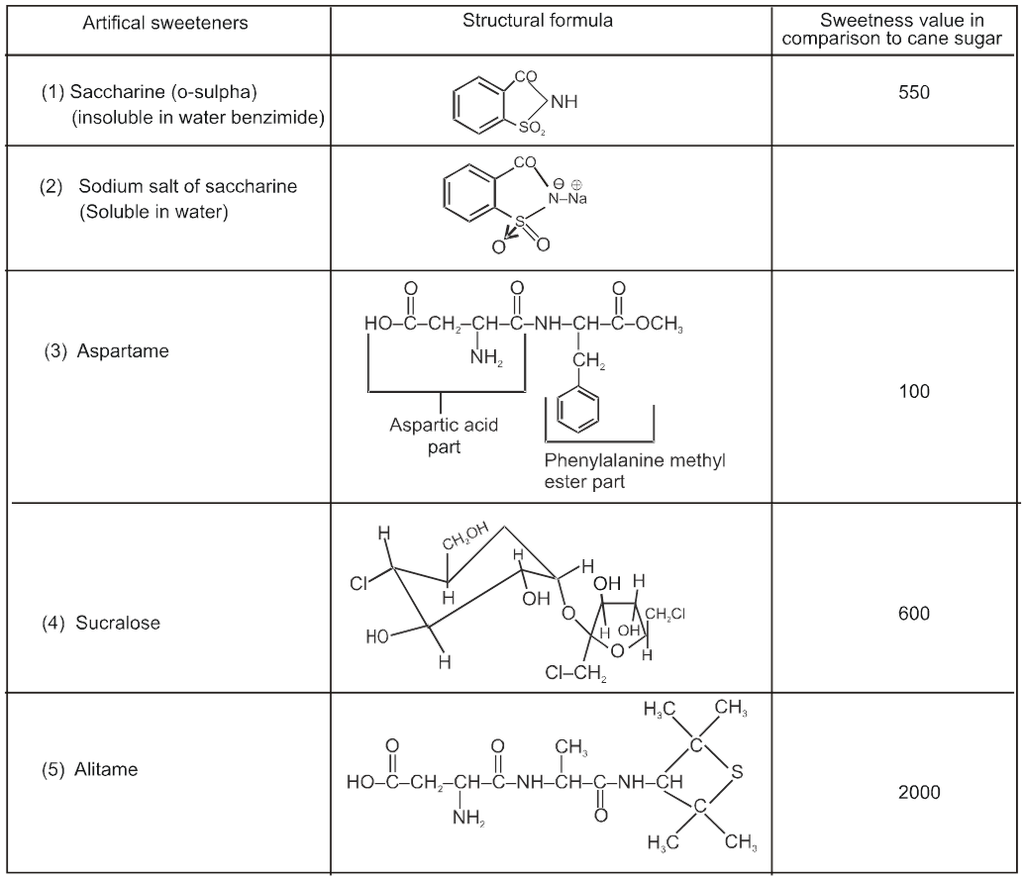
(b) Food Preservatives :
*The chemical which are used to stop undesirable change in food caused by microorganism and save them from spoiling are called preservatives. It reduces (stop the growth) and rate reactions occuring due to bacteria in food).
*The following properties must be present in a preservative :
(i) It should not react with food material.
(ii) It’s effect should be for longer period.
(iii) It should not decrease the quality of food.
(iv) It should not have harmfull effect on the body.
Improtant preservatives are as follows :
(1) Sodium benzoate : It’s 0.06% to 0.1% concentration is used for preservation of fruit juice , jam, jelly, pickles etc.
(2) Parabens : These are alkyl p-hydroxy benzoate and used for preservation of tomato sauce etc.
(3) Sorbates : These are salt of sorbic acid and used for preservation of milk cheese preparation certain meats and fish products. It inhibit the growth of yeast
(4) Propionates : These are ethyl and phenyl ester of propionic acid and used for the preservation of biscuits and baked product, etc from mould fungi etc.
(5) Sodium or potassium metabisulphite (Na2S2O5 or K2S2O5) : It is used as a preservative for food products such as jams, squashes, pickles etc.
(6) Epoxides : Epoxides are gases and preserves low moisture foods like nuts, dried fruits. Epoxides destroy all type of microorganism including spores and viruses.
(7) p-Hydroxy benzoate ester : The methyl, ethyl propyl and heptyl esters of p-hyroxybenzoic acid are used as preservatives in baked foods, soft drinks, beer and syrups.
(c) Antioxidants :
*The chemical substance which reduce the rate of reaction with oxygen in food, thus help in their preservation are called antioxidants.
*They reduce the rate of formation of free radicals responsible for ageing process 2,6 ditertiary butylhydroxy toluene (p-crysol, BHT) and 2-tertiary butyl hydroxy anisole (BHA) are two most familiar antioxidants used.
5. Cleansing Agents :
Soaps and Detergents : *Soaps and detergents are used since long
SOAPS :
Soaps are sodium or potassium salts of long chain fatty acids e.g steric, oleic and palmitic acids. soap containg sodium salts are formed by heating fat (i.e. glyceryl ester of fatty acid) with aqueous sodium hydroxide solution. This reaction is known as SAPONIFICATION. Generally potassium soaps are soft to the skin.
Types of soaps :
There are so many types of soaps due to the using different raw materials
(i) Toilet soaps
(ii) Water floating soaps
(iii) Transparent soaps
(iv) Medicated soap
(v) Shaving soaps
(vi) Loundry soaps
(vii) Soaps chips
(viii) Soap granules
Detergents :
The synthetic products , which like soaps remove dust and grease from a surface are called detergents, since they are not soap but work like a soap so they are also called as soapless soap.
Detergents are mainly sodium salts of either sulphuric or sulphonic acids with long chain hydrocarbons.
eg. : Sodium alkyl sulphate or Sodium alkyl sulphonate
These can be used both in soft and hard water, as they give foam even in hard water
Synthetic detergents are mainly classified into three catagories :
(i) Anionic detergents : These are sodium salt of sulphonated long chain alcohols or hydrocarbons.
eg, : Lauryl alcohol, Lauryl hyrogen sulphate, Sodium lauryl sulphate
In anionic detergents, the anionic part of the molecule is involved in the cleansing action. These are smoothly used for household work and are also used in toothpastes.
(ii) Cationic detergents : These are quatenary ammonium salts of amines with acetates, chlorides or bromides as anion. Cetyltrimethylammonium bromide is a popular cationic detergent.
(iii) Non ionic detergents : These are moslty esters of poly hydroxy alcohols. They are in liquid form, and do not contain any ion in their constiution. One such detergent is formed when stearic acid reacts with polyethyleneglycol.
eg. : Stearic acid, Poly ethyleneglycol
Note : Liquid dish washing detergents are non ionic type.
Difference between soap and detergents
(1) Soaps are salts of weak acid and strong base whereas detergents are salts of strong acid and strong base.
(2) Aqueous solution of soap is basic where as aqueous solution of detergents is neutral.
(3) woolen and silk cloths in which soft fibres are present cannot be washed with soap whereas all type of fabrics can be washed with detergent
(4) Soap cannot work in hard water because soaps are precipitated as insoluble salt by reaction with Ca2+ and Mg2+ ions.
5. Rocket Propellant
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Rocket Propellant
* The propellants are chemical substances which on ignition provide thrust for the rocket to move forward. These substances are called rocket propellants.
(1) Types of rocket propellants : Depending upon the physical state, propellants can be classified as.
(i) Solid propellants : The solid propellants are mixtures of solid fuel and solid oxidiser. These are further divided into to classes.
(a) Composite propellants
(b) Double base propellants
(ii) Liquid propellants : These consist of an oxidizer such as liquid oxygen, nitrogen tetroxide (N2O4) or nitric acid and a fuel such as kerosene, alcohol, hydrazine or liquid hydrogen. These are further classified as,
(a) Monopropellants
(b) Bipropellants
Advantages of Biliquid Propellants over Solid Propellants
The biliquid propellants give higher thrust than solid propellants.
The thrust generated by liquid propellants can be controlled by switching on and off the flow of propellants. On the other hand, the thrust cannot be controlled in solid propellants.
(iii) Hybrid propellants : These are the propellants which consist of solid fuel and a liquid oxidiser. For
example, liquid N2O4 (liquid oxidiser) and acrylic rubber (solid fuel).
(2) Examples of Propellants used in Different Rockets
(i) Saturn booster rocket of American space programme used a mixture of kerosene and liquid oxygen as the propellant in the initial stage where as liquid oxygen and liquid hydrogen were used as propellant in high altitudes.
(ii) Russian rockets such as Proton used a liquid propellent consisting of kerosene and liquid oxygen.
(iii) The Indian satellites SLV-3 and ASLV used composite solid propellants.
(iv) The rocket PLSV will use solid propellant in the first and third stages and liquid propellant in second and fourth stages. The liquid propellant will consist of N2O4 and unsymmetrical dimethyl hydrazine (UDMH) and N2O4 and monomethyl hydrazine (MMH) respectively.
6. Nucleic acids
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Nucleic acids
Nucleic acids were first discovered by a Swiss biochemist, Friedrich Miescher (1869) who called them nuclein due to their acidic nature.Chemical analysis of chromosomes shows presence of two nucleic acids-DNA (Deoxyribo nucleic acid) and RNA (Ribo nucleic acid). Nucleic acid is a macromolecule & consists of many (polymer) monomeric units, called nucleotides. Each nucleotide is composed of a nucleoside and a phosphate group. Thus nucleotide is a phosphoric ester of nucleoside.
Each nucleoside consists of sugar molecule and a nitrogenous base. The relationship can be shown as given below.
Nucleic acid =many nucleotides
Nucleotide =nucleosides + phosphate
Nucleoside =sugar + nitrogenous base
Thus nucleotide =phosphate + sugar+ nitrogenous base
Nucleic acids bear different components that are briefly discussed below.
1. Phosphoric acid : The acidic nature of nucleic acids is due to the presence of phosphoric acid. Sugar of nucleoside combines with phosphoric acid by a phosphodiester bond formed at 5th or 3rd carbon of the sugar.
2. Sugar : It is a five carbon (pentose) sugar. There are two types of pentose sugars-ribose and deoxyribose. Deoxyribose sugar has one oxygen atom less at second carbon. Ribose sugar is present in RNA while deoxyribose sugar occurs in DNA.
3. Nitrogenous bases : they have two catagories
(a) Pyrimidine:- It includes cytosine, thymine and uracil. Pyrimidine bases are made of only one ring of carbon.
(b) purine:- It includes Adenine and guanine. Purine bases are made of two ring of carbon and nitrogen bases of DNA contains adenine, guanine, cytosine and thymine. In RNA, uracil is present in place of thymine.
Many nucleotide monomer units join one another to give rise to polynucleotide chain.
The two adjacent nucleotides are joined by formation of phosphodiester bond (a bond that involves two ester bonds). A polynucleotide chain is often written as 5’p 3’OH. This indicates that it is a dinucleotide with phosphate group (p) attached to the 5th carbon of terminal nucleotide and hydroxyl group (OH) is present at 3rd carbon of basal nucleotide.
Structure of DNA (Deoxyribonucleic acid) :
J.D. Watson and F.H.C. Crick (1953)
Proposed double helical structure of DNA based on the results of M.H.F.Wilkins and co-workers. All these three persons were awarded Nobel Prize in 1962 for this work.
The following are some of the characteristic features of double helical structure of DNA.
(i) Each nucleotide consists of sugar, phosphate and a nitrogenous base. Many such nucleotides are linked by phosphodiester bonds to form a polynucleotide chain or strand.
(ii) Phospho diester bonds are formed between 5’carbon of sugar of one nucleotide and 3' carbon of sugar of the next nucleotide.
(iii) Nitrogenous base is attached to1’ carbon of sugar. At this place purine base is attached by its 5' position and pyrimidine by its 3' position.
(iv) Polynucleotide strand is made of backbone of sugar and phosphate forming its long axis and bases at right angles to it.
Chargaff (1950)
Made observations on the base and other contents of DNA. These observations or generalizations are called Chargaffs rule.
(1) Purine and pyrimidine base pairs are in equal amount, that is, adenine + guanine =thymine + cytosine.
(2) Molar amount of purine-adenine is always equal to the molar amount of pyrimidine-thymine. Similarly, guanine is equalled by cytosine.
(3) Sugar deoxyribose and phosphate occur in equimolar proportions.
(4) The ratio of A + T/G + C is constant for a species.
(5) Chargaffs rule states that in natural DNAs the base ratio A/T is always close to unity and the G/C ratio also to always close to unity. It indicate that A always pairs with T and G pairs with C. A and T, G and C, therefore,are complementary base pairs.
(6) Thus, if one DNA strand has A, the other would have T and if it has G, the other, would have C. Therefore, if the base sequence of one strand is CAT TAG GAC, the base sequence of other strand would be GTA ATC CTG. Hence, the two poly nucleotide strands are called complementary to one another.
(7) Two such complementary strands are joined with each another by hydrogen bonds between their complementary nitrogenous bases. There are three hydrogen bonds between cytosine and guanine and two hydrogen bonds between adenine and thymine.
(8) The two polynucleotide chains are helically coiled around the same axis in such a way that these can separate from one another only by uncoiling. Helical coiling is supposed to be right handed. Such a form of DNA is now called B-DNA
(9) The two chains or strands are antiparallel, i.e., they run in opposite directions in relation to their sugar molecules. Their 5’p 3' OH phosphodiester linkages are in opposite directions
(10) Double stranded DNA molecule has a diameter of 20Aº.
(11) The helix makes one complete turn every 34 Aº along its length.
(12) There are 10 nucleotides per turn of helix.Thus the distance between two neighbouring base pairs is 3.4 A
RNA :
RNA is ribonucleic acid formed in the nucleus and is found in the cytoplasm of the cell.
Types of RNA and their functions :
(1) r-RNA (Ribosomal – RNA) :
It is found in the ribosomes and it is usually associated with protein to form the ribosomes. It is synthesised in the nucleus by DNA. It is single stranded, comprising about 80% of the total RNA. It is metabolically stable.
Functions : (i) This forms the site for protein synthesis.
(ii) It is also supposed to help the binding of m-RNA to the ribosomes during protein synthesis.
(2) m-RNA (Messenger RNA) :
It is carries the genetic message code from the D.N.A. to ribosomes. It is produced by the DNA ; m-RNA is also single stranded and constitutes about 15% of total RNA.
Functions : It carries the genetic information from DNA to the ribosomes where protein is synthesised.
(3) t-R.N.A (Transfer–RNA) :
It is synthesised in the nucleus by the DNA. It is also called soluble RNA. It is single stranded. There are 20 different kinds of t-RNA and each type has a specificity for a particular amino acid. It constitutes about 5% of total RNA. It has very short life.
Functions : It carries amino acids from different parts of cytoplasm to the ribosomes during protein synthesis.
9. Metal Carbonyls
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Metal Carbonyls
Compounds of metals with CO as a ligand are called metal carbonyls.
They are of two types.
(a) Monomeric : Those metal carbonyls which contain only one metal atom per molecule are called monomeric carbonyls. For examples :[Ni(CO)4] (sp3, tetrahedral) ; [Fe(CO)5 ] (dsp3, trigonal bipyramidal)
[Cr(CO)6] (d2 sp3, octahedral) ; [V(CO)6] (d2 sp3, octahedral, only carbonyl which is paramagnetic having one unpaired electron. This is least stable among all the four carbonyls)
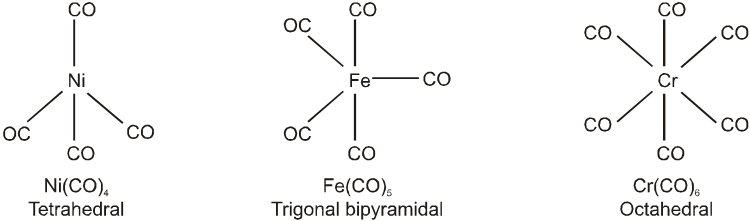
(b) Polymeric : Those metal carbonyl which contain two or more than two metal atom per molecule and they have metal-metal bonds are called polymeric carbonyl. For example : Mn2 (CO)10 , Co2(CO)8, etc.
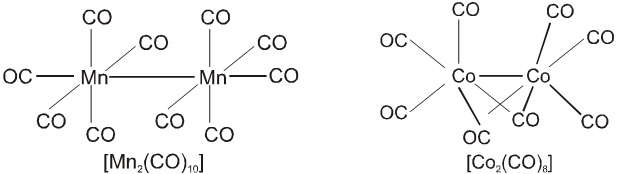
The metal–carbon bond in metal carbonyls possess both s and p character. The M—C s bond is formed by the donation of lone pair of electrons on the carbonyl carbon (CO is a weak base) into a vacant orbital of the metal. The M — C p bond is formed by the donation of a pair of electron from a filled d orbital of metal into the vacant antibonding p* orbital of carbon monoxide. Thus carbon monoxide acts as s donor (OC ¬ M) and a p acceptor (OC ¬ M), with the two interactions creating a synergic effect which strengthens the bond between CO and the metal as shown in figure.
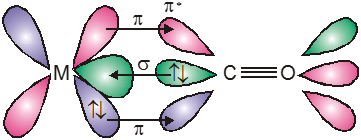
Synergic bonding
10. STABILITY OF COORDINATION COMPOUNDS
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Stability of coordination compounds :
The stability of a coordination compound [MLn] is measured in terms of the stability constant (equilibrium constant) given by the expression,
bn = [MLn]/[M(H2O)n][L]n
for the overall reaction :
M(H2O)n + nL ![]() MLn + nH2O
MLn + nH2O
By convention, the water displaced is ignored, as its concentration remains essentially constant. The above overall reaction takes place in steps, with a stability (formation) constant, K1, K2, K3, ...... Kn for each step as represented below :
M(H2O)n + L ![]() ML(H2O)n–1 + H2O
ML(H2O)n–1 + H2O
K1 = [ML(H2O)n–1] / {[M(H2O)n][L]}
MLn–1 (H2O) + L ![]() MLn + H2O
MLn + H2O
Kn = [MLn] / {[MLn–1 (H2O)] [L]}
M(H2O)n + nL![]() MLn + nH2O
MLn + nH2O
bn = K1 x K2 x K3 x ........ x Kn
bn, the stability constant, is related to thermodynamic stability when the system has reached equilibrium. Most of the measurements have been made from aqueous solutions, which implies that the complex is formed by the ligand displacing water from the aqua complex of the metal ion. Ignoring the charge and taking L as an unidentate ligand, the stepwise formation of the complex is represented as shown above. K1, K2, K3 ..... Kn representing the stepwise stability (or formation) constants.
The above is thermodynamic stability criteria, there can be another kind of stability called Kinetic Stability, which measures the rate of ligand replacement.
Some important generalisation regarding stability constants :
For a given metal and ligand the stability is generally greater when the charge on the metal ion is greater. Thus, stability of coordination entities of ions of charge 3+ is greater than the entities of 2+ ions. Further, for the divalent ions of the first row transition elements, irrespective of the ligand involved, the stabilities vary in the Irving-Williams order :
MnII < FeII < CoII < NiII < CuII > ZnII
This order is according to the size of the ions, smaller the size of the ion or greater the charge density on the metal greater is the stability of the complex.
In F¯, Cl¯, Br¯, I¯ ; F¯ forms strongest complexes due to small size & hence high charge density.
The stability also depends on the formation of chelate rings. If L is an unidentate ligand and L–L, a didentate ligand and if the donor atoms of L and L–L are the same element, then L–L will replace L. The stabilisation due to chelation is called the chelate effect. It is of great importance in biological systems and analytical chemistry. The chelate effect is maximum for the 5- and 6-membered rings. In general, rings provide greater stability to the complex.
11. Isomerism in coordination compounds :
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Isomerism in coordination compounds :
(1) Structural isomerism :
The structural isomerism arises due to different lignads within the coordination sphere.
(A) Ionisation Isomerism
This type of isomerism occurs when the counter ion in a coordination compound is itself a potential ligand and can displace a ligand which can then become the counter ion. For example, following complexes show ionisation isomerism.
[Co(NH3)5SO4]NO3 and [Co(NH3)5NO3]SO4
[Co(NH3)4(NO2)Cl]Cl and [Co(NH3)4Cl2]NO2.
[Co(NH3)4(H2O)Cl]Br2 and [Co(NH3)4BrCl]Br.H2O. [Also an example of hydrate isomers.]
[Pt(NH3)4Cl2]Br2, and [Pt(NH3)4Br2]Cl2.
[CoCl(en)2(NO2)]SCN, [Co(en)2(NO2)SCN]Cl and [Co(en)2(SCN)Cl]NO2
(B) Solvate/Hydrate Isomerism :
It occurs when water forms a part of the coordination entity or is outside it. This is similar to ionisation isomerism. Solvate isomers differ by whether or not a solvents molecule is directly bonded to the metal ion or merely present as free solvent molecules in the crystal lattice. For example, CrCl3 . 6H2O exists in three distinct isomeric forms : [Cr(H2O)6]Cl3, violet ; [CrCl(H2O)5]Cl2.H2O, blue green : [CrCl2(H2O)4]Cl.2H2O, dark green.
(C) Linkage isomerism :
In some ligands, like ambidentate ligands, there are two possible coordination sites. In such cases, linkage isomerism exist. e.g., NO2 group can be bonded to metal ions through nitrogen (–NO2) or through oxygen (–ONO). SCN too can be bonded through sulphur (–SCN) thiocyanate or through nitrogen (–NCS) isothiocyanate.
For example : [Co ONO (NH3)5] Cl2 & [Co NO2 (NH3)5] Cl2 .
(D) Coordination Isomerism
Coordination compounds made up of cationic and anionic coordination entities show this type of isomerism due to the interchange of ligands between the cation and anion entities. Some of the examples are :
(i) [Co(NH3)6][Cr(CN)6] and [Cr(NH3)6](Co(CN)6]
(ii) [Cu(NH3)4](PtCl4] and [Pt(NH3)4][CuCl4]
(iii)[Co(NH3)6][Cr(SCN)6]and[Cr(NH3)4(SCN)2](Co(NH3)2(SCN)4]
(iv) [Pt(NH3)4][PtCl6] and [Pt(NH3)4Cl2][PtCl4]
Such isomers are expected to have significant differences in their physical and chemical properties.
(2) Stereoisomerism
The isomers in which atoms are bonded to each other in the same order but that differ in the arrangement of these atoms in the space are called as stereoisomers and the phenomenon as stereoisomerism.
(A) Geometrical Isomerism
This type of isomerism arises in heteroleptic complexes due to different possible geometric arrangements of the ligands. Geometrical isomerism is common among coordination compounds with coordination numbers 4 and 6.
(i) Coordination Number Four :
(a) Tetrahedral Complex :
The tetrahedral compounds can not show geometrical isomerism as we all know that all four positions are equivalent in tetrahedral geometry.
(b) Square Planar Complex :
In a square planar complex of formula [Ma2b2] [a and b are unidentate], the two ligands ‘a’ may be arranged adjacent to each other in a cis isomer, or opposite to each other in a trans isomer as depicted.
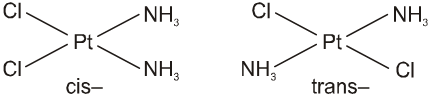
Geometrical isomers (cis and trans) of Pt(NH3)2Cl2 .
Square planar complex of the type Ma2bc (where a,b,c are unidentates) shows two geometrical isomers.

Square planar complex of the type Mabcd (where a,b,c,d are unidentates) shows three geometrical isomers.

Example is [Pt(NH3)BrCl(py)]. three isomers of the complex [Pt(NH3)(NH2OH)(py)(NO2)]+ have been isolated and identified.
Square planar complex of the type M(AB)2 (where AB are unsymmetrical bidentates) shows two geometrical isomers. Example is [Pt(gly)2] in which gly is unsymmetrical ligand.
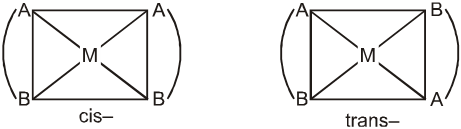
Similarly, M(AB)(CD) also shows two geometrical isomers.
Note : M(AA)2, (where AA are symmetrical bidentates) does not show geometrical isomerism. e.g., [Cu(en)2]2+ [Pt(ox)2]2–, etc.
(ii) Coordination Number Six :
Geometrical isomerism is also possible in octahedral complexes.
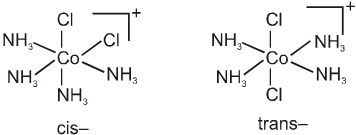
Geometrical isomers (cis and trans) of [Co(NH3)4Cl2]+
Number of possible isomers and the spatial arrangements of the ligands around the central metal ion for the specific complexes are given below.
(I) Complexes containing only unidentate ligands
(a) Ma2b4 – 2
(b) Ma4bc – 2
(c) Ma3b3
Complexes of the formula Ma3b3, where a and b are monodentate ligands, may show two isomeric forms called fac– and mer–. Facial isomers have three identical ligands on one triangular face where as meridional isomers have three identical ligands in a plane bisecting the molecule. Similar isomers are possible with some chelating ligands.
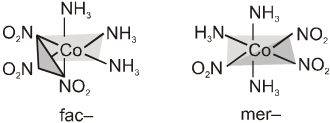
The facial(fac) and meridional(mer) isomers of [Co(NH3)3(NO2)3].
Unsymmetrical bidentate ligands also show fac-mer isomerism
(d) Ma3b2c – 3
(e) Ma3bcd – 4
(f) Ma2b2c2 – 5
(g) Ma2b2cd – 6
(h) Ma2bcde – 9
(i) Mabcdef, [Pt(py)(NH3)(NO2)(Cl)(Br)(I)] – 15
(II) Compounds containing bidentate ligand and unidentate ligands.
(i) M(AA)a3b – Two geometrical isomers are possible.
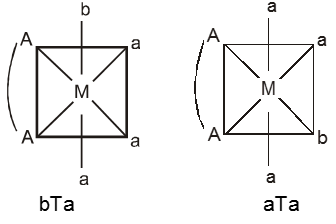
(ii) M(AA)a2b2 – Three geometrical isomers are possible.
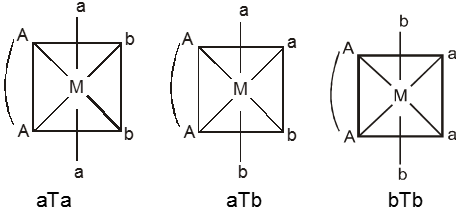
(B) Optical Isomerism :
A Coordination compound which can rotate the plane of polarised light is said to be optically active. When the coordination compounds have same formula but differ in their ability to rotate directions of the plane of polarised light are said to exhibit optical isomerism and the molecules are optical isomers. Optical isomers are mirror images that cannot be superimposed on one another. These are called as enantiomers. The molecules or ions that cannot be superimposed are called chiral. This is due to the absence of elements of symmetry in the complex. The two forms are called dextro(d) and laevo(l) depending upon the direction they rotate the plane of polarised light in a polarimeter (d rotates to the right,l to the left).
(a) Octahedral complexes :
Optical isomerism is common in octahedral complexes involving didentate ligands. For example, [Co(en)3]3+ has d and l forms as given below
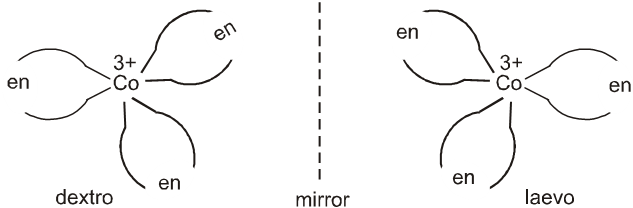
Cis-isomer of [PtCl2(en)2]2+ show optical isomerism as shown below because of the absence of plane of symmetry as well as centre of symmetry.
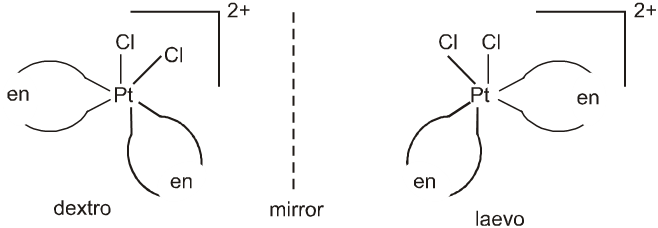
But trans isomer of [PtCl2(en2)]2+ does not show optical isomerism.
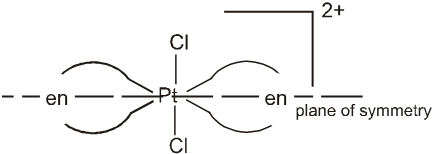
cis-[Co(NH3)2Cl2(en)]+ can show optical isomerism due to the absence of plane of symmetry as well as centre of symmetry.
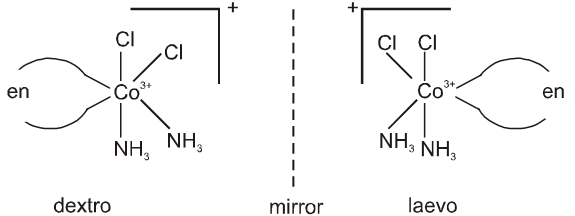
(b) Tetrahedral Complexes.
Optical isomerism is expected in tetrahedral complexes of the type [Mabcd] analogous to tetrahedral carbon atom.
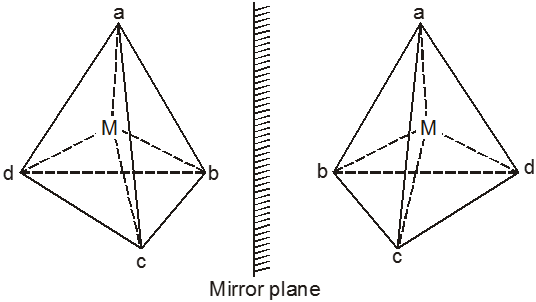
(c) Square Planar Complexes.
Square planar complexes are rarely found to show the optical isomerism. The plane formed by the four ligating atoms and the metal ion is considered to be a mirror plane and thus prevents the possibility of chirality.
12. Applications of coordination and organometallic compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Applications of coordination and organometallic compounds
(i) Coordination compounds are of great importance in biological systems. Example being – chlorophyll (the green pigment in plants); haemoglobin (the red pigment of blood, which acts as oxygen carrier) along with myoglobin (which stores oxygen and is a regulator of respiration); Vitamin B12, cyanocobalammine, the anti-pernicions anaemia factor. All of these, respectively, are the coordination compounds of magnesium, iron and cobalt with the macrocyclic porphyrin and corrin ligands.
(ii) There are many examples of the use of coordination compounds in qualitative and quantitative chemical analysis. The familiar colour reactions given by metal ions with a number of ligands (especially the chelating ligands), as a result of formation of coordination entities, form the basis for their detection and estimation by classical and instrumental methods of analysis. Familiar examples of such reagents are : ethylenediaminetetraaceticacid (EDTA), dimethylglyoxime, a-nitroso b-naphthol, cupron, etc.
(iii) Some important extraction processes of metals, like those of extraction of silver and gold, make use of complex formation. Gold, for example, combines with cyanide in the presence of oxygen and water to form the coordination entity [Au(CN)2]– in aqueous solution. Gold can be precipitated from this solution by the addition of Zinc.
(iv) Purification of metals can be achieved through formation and subsequent decomposition of their coordination compounds. For example, impure nickel is converted to [Ni(CO)4], which is decomposed to yield pure nickel.
(v) Organometallic compounds are used as catalysts. These catalysts are either of the homogeneous type (soluble in the reaction medium) or of the heterogeneous type (insoluble in the reaction medium). The catalysed polymerisation of alkenes at atmospheric pressure and ambient temperature using Ziegler-Natta catalyst (titanium tetrachloride plus triethylaluminium) is one of the important discoveries of organometallic chemistry. The first effective homogeneous catalyst chloridotris(triphenylphosphine) rhodium(I), [RhCl(PPh3)3] for hydrogenation was given by Wilkinson.
(vi) Tetra ethyl lead (TEL) is used as antiknock compound in gasoline.
(vii) Articles can be electroplated with silver and gold much more smoothly and evenly from solutions of the complexes, [Ag(CN)2]– and [Au(CN)2]– than from a solution of simple metal ions.
(viii) In black and white photography, the developed film is fixed by washing with hypo solution which dissolves the undecomposed AgBr to form a complex ion, [Ag(S2O3)2]3–.
(ix) The chelate therapy is now a day is used in medicinal chemistry.
(a) Excess of copper and iron are removed by the chelating ligands D-penicillamine and desferrioxime B via the formation of coordination compound in plant/animal systems.
(b) EDTA is used in the treatment of lead poisoning.
(c) Coordination compounds of platinum like cis-platin and related compounds effectively inhibit the growth of tumours.
8. Carbon tetrachloride (pyrene)
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Carbon tetrachloride (pyrene)
Preparations & reactions :
- CCl4 is stable to heat and its vapours do not catch fire thus it is used as fire extinguisher.
- CCl4 used as medicine for elimination of hook worms.
Freons (Polychlorofluoro alkane)
Nomenclature of Freons :
Freon is expressed as
Freon - cba
Where
a – [Number of F-atoms]
b – [1 + Number of H–atoms]
c – [Number of C-atoms –1]
Example : C2Cl4F2 is expressed as Freon-112
a = 2 ; b = 1 + 0 ; c = 2 – 1
cba = 112
- Freon is odourless, non-corrosive, non-toxic gas. It can easily be liquefied hence widely used as refrigerant.
9. Alchols and Ethers – Introduction and Classification
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Alchols and Ethers – Introduction and Classification
Alcohols :
Alcohols have sp3 hybridized oxygen atoms, but the C – O – H bond angle in methanol (108.9°) is considerably larger than the H – O – H bond angle in water (104.5°) because the methyl group is much larger than a hydrogen atom.
The bulky methyl group counteracts the bond angle compression caused by oxygen’s nonbonding pairs of electrons. The O – H bond lengths are about the same in water and methanol (0.96 Å), but the C – O bond is considerably longer (1.4 Å), reflecting the larger covalent radius of carbon compared to hydrogen.

Classification of alcohols:
(a) Alcohols may be classified as mono–, di–, tri- or polyhydric alcohols depending on whether they contain one, two, three,.............hydroxy group.
Monohydric : Contains one –OH group (C2H5OH ; CH3–CH2–CH2–OH)
Dihydric : Contains two –OH group ![]() (Glycol)
(Glycol)
Trihydric (Polyhydric): Contains three or more than three –OH groups ![]() (Glycerol)
(Glycerol)
(b) Allylic alcohols: CH2=CH–CH2–OH ; 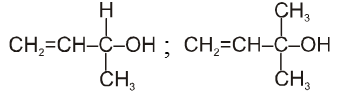 ;
;
(c) Vinylic Alcohol: CH2=CH–OH (unstable at room temperature)
(d) Benzylic alcohols:
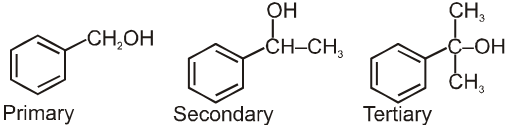
Ethers :
Like water, ethers have a bent structure, with an sp3 hybrid oxygen atom giving a nearly tetrahedral bond angle.

Bonding in ethers is readily understood by comparing ethers with water and alcohol. Van der Waals strain involving alkyl groups causes the bond angle at oxygen to the larger in ethers than in alcohol, and larger in alcohols than in water. An extreme example is di-tert-butyl ether, where steric hindrance between the tert-butyl groups is responsible for a dramatic increase in the C – O – C bond angle.
Classificiation of ethers :
(a) Simple or symmetrical : If the alkyl or aryl groups attached to the oxygen atom are the same.
CH3OCH3, C2H5OC2H5
(b) Mixed or unsymmetrical : If the two groups attached to the oxygen atom are different.
C2H5OCH3, C2H5OC6H5.
General Reactions of alkyl halides, alcohols & ethers :

10. Nucleophilic substitution (SN) reaction of alcohols
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Nucleophilic substitution (SN) reaction of alcohols
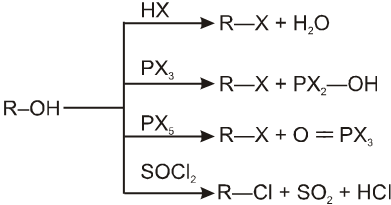
In presence of HX :
R–OH + HX RX + H2O
This is a nucleophilic substitution (SN1/SN2)
Mechanism :
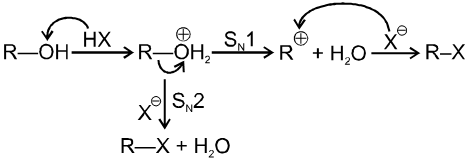
Note : Mainly - unsubstituted 1º alcohol give SN2 reaction with HX.
Ex.
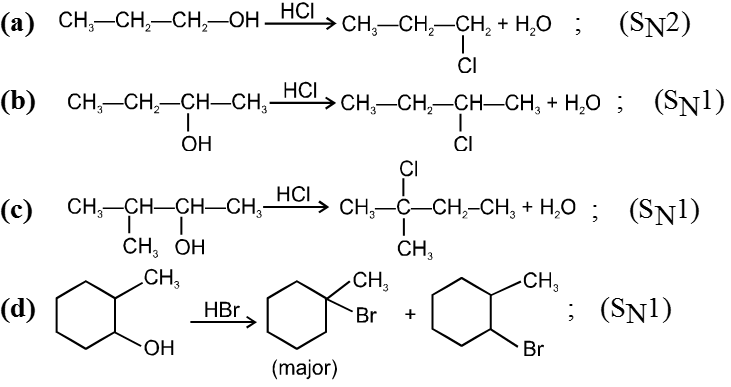
Reactivity of HX is HI > HBr > HCl > HF.
Finkelstein reaction :
R—OH + HI R—I  R—H + I2 (reduced) [Final product is Alkane (R-H) not R-I]
R—H + I2 (reduced) [Final product is Alkane (R-H) not R-I]
This is a problems that is why iodides are best prepared through halogen exchange reaction. It is also known as Finkelstein reaction. In this reaction R—Cl and R—Br is treated with sodium iodide in acetone.
![]()
![]()
NaI is soluble in acetone. In this reaction acetone is used because sodium iodide is soluble in acetone but NaBr and NaCl are insoluble so precipitated out. This eliminates any possibility of reverse reaction.
It is an SN2 reaction therefore only 1ºRX and 2ºRX is used.
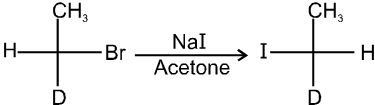
Lucas reagent :
The 1 : 1 mixture of anhydrous ZnCl2 : HCl (conc.) is called Lucas reagent which is used to distinguish between 1º, 2º and 3º alcohols.
![]()
Mechanism :
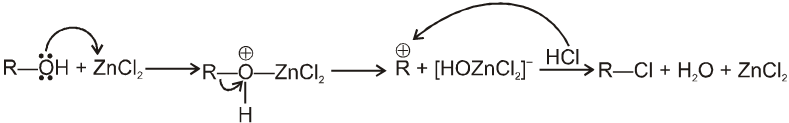
ZnCl2 increases the rate of reaction by making –OH group into a much better leaving group just through complexation.
Reactivity of alcohols is 3ºROH > 2ºROH > 1ºROH
Ex.
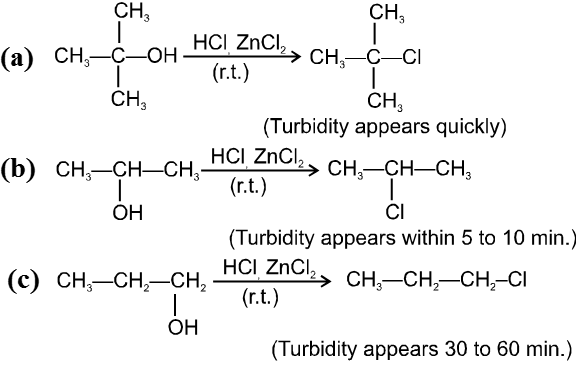
1.4 In presence of PX3 :
R–OH  R–Br
R–Br
Mechanism :

2R—OH + HOPBr2 2R–Br +
1.5 In presence of PX5 :

Both the PX3 & PX5 proceeds via SN2 pathways, No.rearrangement occurs.
Only 1ºR—OH and 2ºR—OH undergo this types of reaction. 3º R—OH undergoes elimination reaction.
Ex. 
1.6 In presence of SOCl2 (Thionyl chloride) :
R—OH R—Cl + SO2 (SN2) ; R—OH
R—Cl + SO2 (SN2) ; R—OH  R—Cl + SO2 (SNi)
R—Cl + SO2 (SNi)
(A) Mechanism of SOCl2 with pyridine :

SOCl2 with pyridine undergo SN2 reaction so inversion of configuration is obtained in product.
(B) Mechanism of SOCl2 without pyridine : (Darzen method)

SOCl2 without pyridine or base undergo SNi reaction, so retention of configuration is obtained in product.
Ex. (a) 
(b) 
1.7 Victor Mayer test :
1° Alcohol%

2° Alcohol%

3° Alcohol %

11. E1 Reaction of alcohols
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
E1 Reaction of alcohols
For E1 mechanism reagents are H3PO4 /D, H2SO4 / 160º
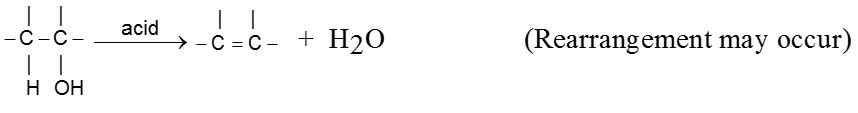
Mechanism :
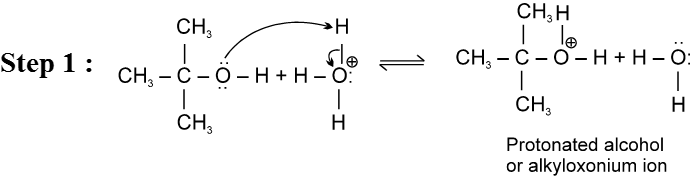
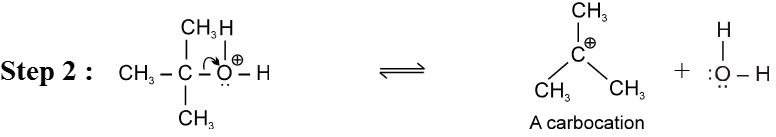
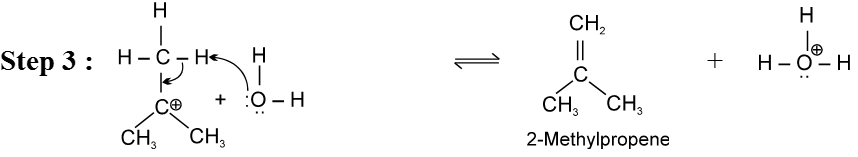
- Reactivity of ROH is 3° > 2° > 1°.
Ex.
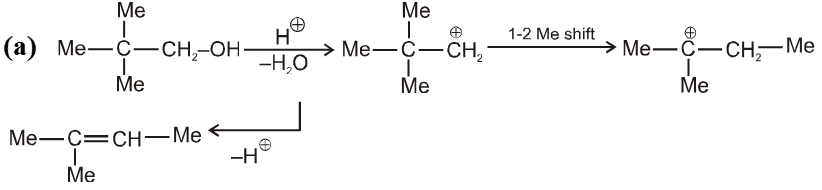

12. E2 Reaction of alcohols
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
E2 Reaction of alcohols
Alcohols gives E2 reaction with Al2O3, P2O5, ThO2, WO3
![]()
- With Al2O3 and P2O5 Saytzeff alkene is major product but with ThO2 and WO3 Hofmann alkene is major product.
Pinacol-Pinacolone rearrangement :
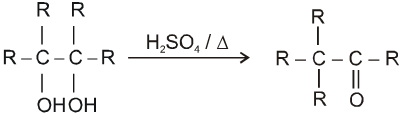
Mechanism :
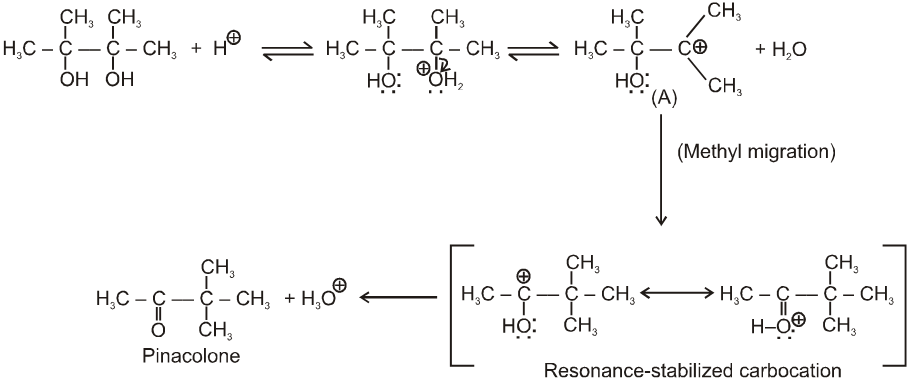
Note : For unsymmetrical diol that –OH group will be replaced first which will generate stable carbocation as below .
Ex.
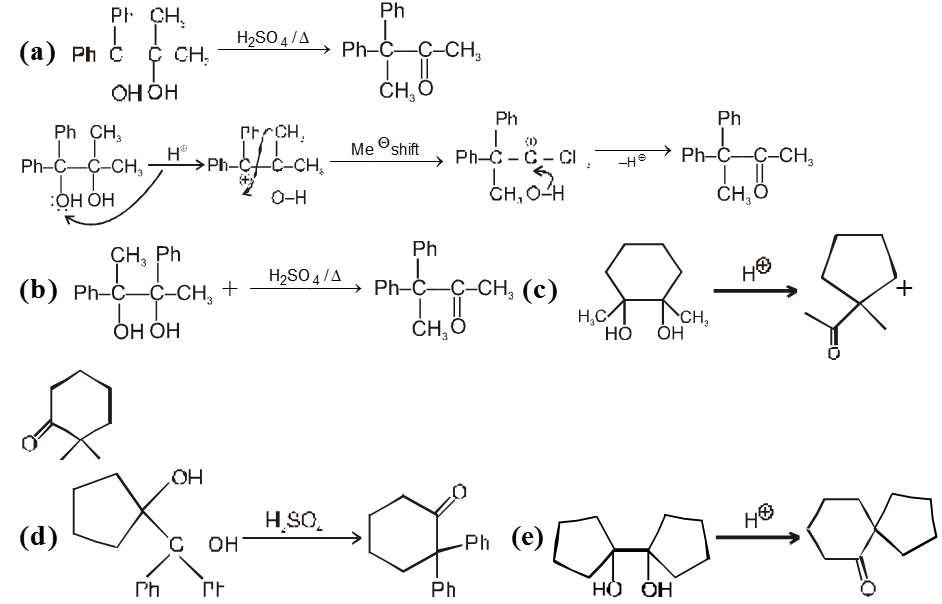
13. Ethers
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Ethers
Preparation of ether :
(i) From Wiliamsons synthesis method :
![]()
Ex.
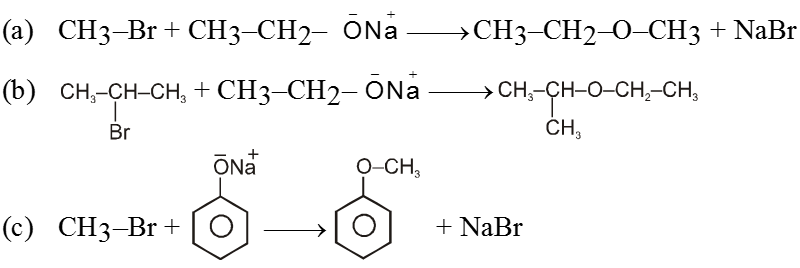
(ii) By using dry Ag2O :
![]()
Ex.
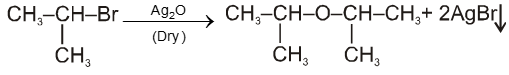
(iii) From alcohols :
![]()
(Trace)
- By intermolecular dehydration of alcohols ethers can be prepared in presence of trace amount of H2SO4.
Mechanism :
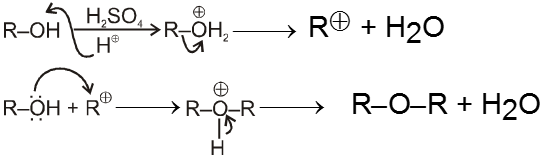
Purification of ethers :
On standing in contact with air they form unstable peroxide (R2O ® O) which is explosive in nature. Peroxide is formed at the carbon atom next to the etherial oxygen.
- The presence of peroxide in ether can be identified on shaking it with freshly prepared ferrous sulphate solution followed by addition of potassium thiocyanate. This result into red colour of ferric thiocyanate. During the reaction peroxide oxidises Fe2+ to Fe3+ which reacts with thiocyanate ion to give red colour of ferric thiocyanate.
![]()
(Red colour)
- Ethers are purified from peroxide before use. The peroxide from ether can be removed by treating ether with KI solution where peroxide is reduced to ether and iodide ion is oxidised to I2 molecule.
- Peroxide formation can be checked by adding few ammount of Cu2O in ether
- Ether is lewis base hence forms co-ordinate bond with lewis acid
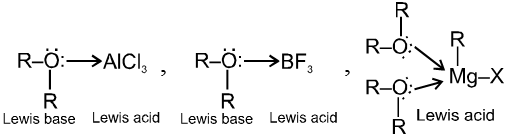
Chemical properties of ethers :
(i) Reaction with HI :
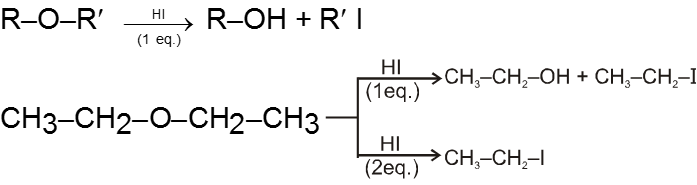
- Those protic acids whose conjugate base is a good nucleophile leads to cleavage of ethers. HI is such a very prominent acid of this kind.
- In most cases of unsymmetrical ethers 1 eq. of HI leads to give a mixture of four products in which it is not possible to predict the major product. However if 2 eq. of HI is used then two iodides are obtained.
Ex.
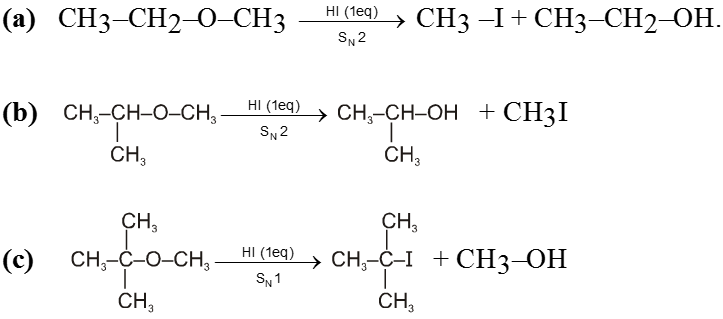
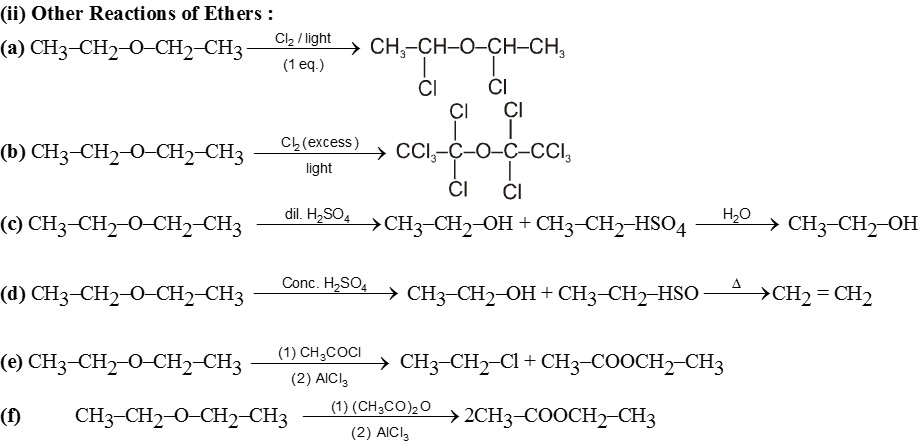
12. Carboxylic Acid Derivatives
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Carboxylic Acid Derivatives
Closely related to the carboxylic acids and to each other are a number of chemical families known as functional derivatives of carboxylic acids : acid chlorides, anhydrides, amides, and esters, These derivatives are compounds in which the —OH of a carboxyl group has been replaced by —CI,—OOCR,
—NH2, or —OR`.
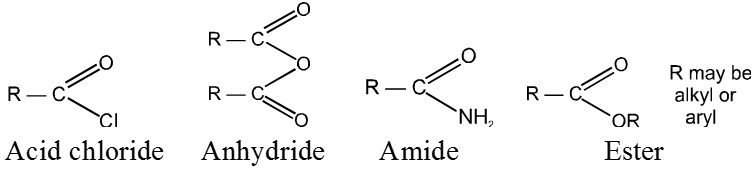
They all contain the acyl group, ![]()
Like the acid to which it is related, an acid derivative may be aliphatic or aromatic, substituted or unsubstituted; whatever the structure of the rest of the molecule, the properties of the functional group remain essentialy the same.
Characteristic reaction of acid deerivatives (Nucleophilic acyl substitution) :
Nucleophilic acyl substitution usually takes place by an addition-elimination mechanism.The incoming nucleophile adds to the carbonyl to form a tetrasubstituted intermediate with a tetrahedral carbon.
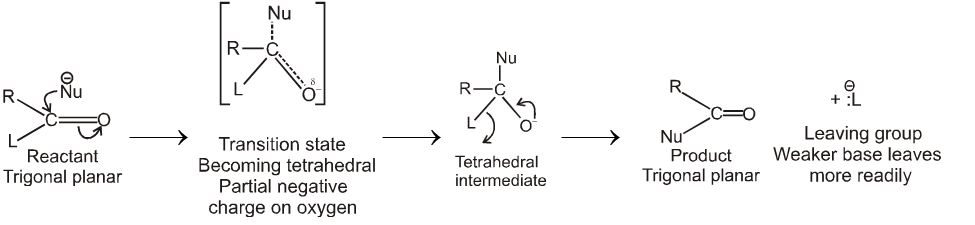
The tetrahedral intermediate formed when a nucleophile attacks the carbonyl carbon of a carboxylic acid derivative is not stable and cannot be isolated.
A pair of nonbonding electrons on the oxygen reforms the bond, and either  or
or  is eliminated with its bonding electrons. Whether or is eliminated depends on their relative basicities. The weaker base is preferentially eliminated because the weaker the base, the better it is a leaving group.
is eliminated with its bonding electrons. Whether or is eliminated depends on their relative basicities. The weaker base is preferentially eliminated because the weaker the base, the better it is a leaving group.
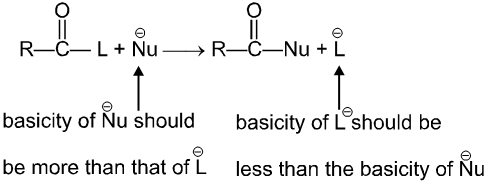
Thus carboxylic acid derivative will undergo a nucleophilic acyl substitution reaction provided that the incoming nucleophile is a stronger base than the group that is to be replaced. If the incoming nucleophile and the group attached to acyl group in the starting material have similar basicities, the tetrahedral intermediate can expect either group with similar ease. A mixture of starting material and substitution product will result.
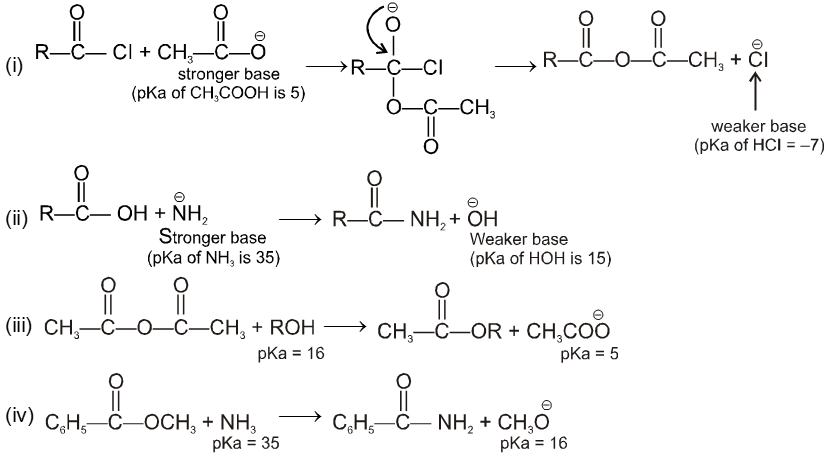
Condition for acyl nucleophilic substitution reaction :
![]()
(i) L must be better leaving group than ![]() , i.e., basicity of Nu should be more than that of
, i.e., basicity of Nu should be more than that of
(ii) ![]() must be a strong enough nucleophilic to attack RCOL.
must be a strong enough nucleophilic to attack RCOL.
(iii) Carbonyl carbon must be enough electrophilic to react with ![]() .
.
Reactivity order : 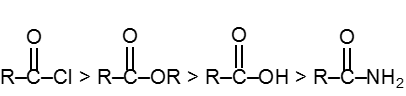
(A) Acid halides :
Methods of preparation of Acyl halides :
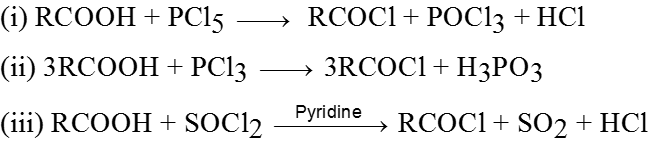
Ex. 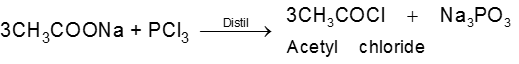
Ex. 
Chemical Reactions :

(B) Acid amides :
Methods of preparation of acids amides :
(1) By reaction of esters with ammonia and amines :
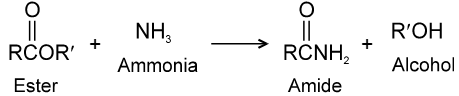
Ammonia is more nucleophilic than water, making it possible to carry out this reaction using aqueous ammonia.
(2) From acid halides :
RCOCl + 2NH3![]() RCONH2 + NH4Cl
RCONH2 + NH4Cl
(3) From anhydride :
(RCO)2O + 2NH3 ![]() RCONH2 + RCOONH4
RCONH2 + RCOONH4
(4) From ammonium salt of carboxylic acid :
RCOONH4 ![]() RCONH2 + H2O
RCONH2 + H2O
CH3COONH4 ![]() CH3CONH2
CH3CONH2
(5) From cyanides :
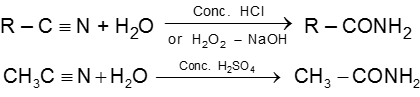
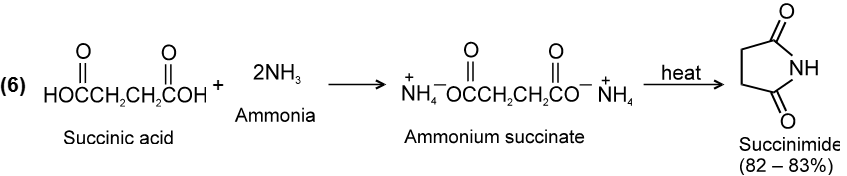
Chemical Reactions :
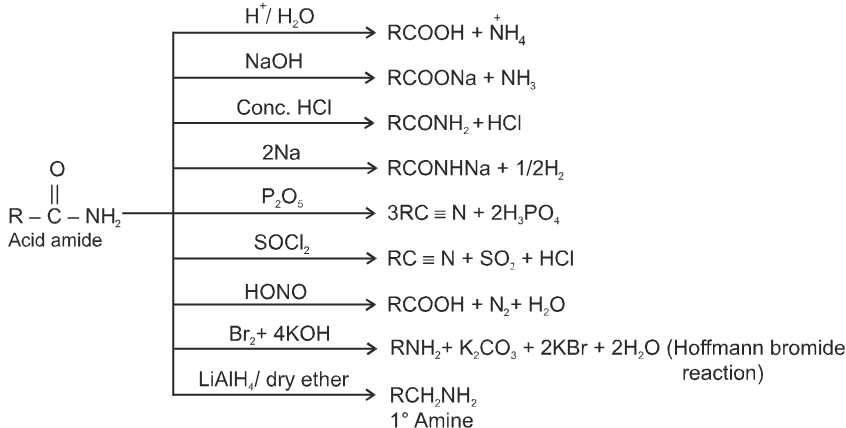
(C) Esters :
Methods of Preparation :
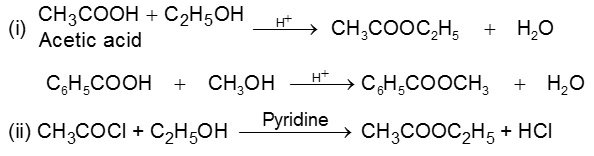
Alcohols react with acyl chlorides by nucleophilic acyl substitution to yield esters. These reactions are typically performed in the presence of a weak base such as pyridine.
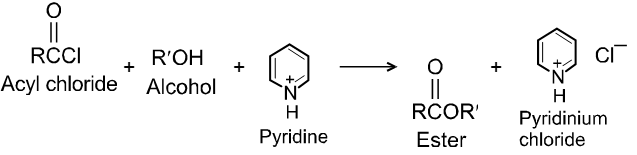
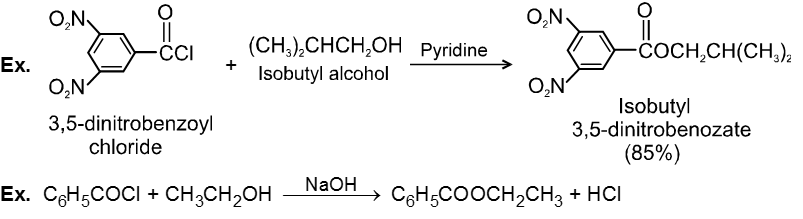
Chemical Reactions :
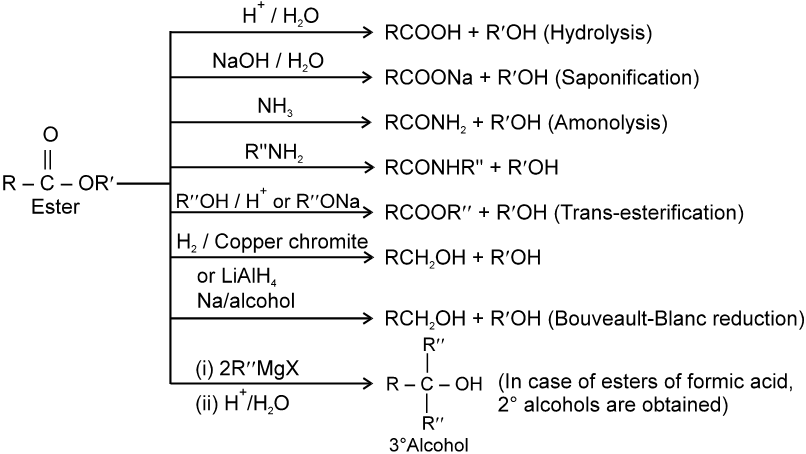
(D) Acid anhydrides :
Methods of Preparation of acid anhydrides :
(1) From carboxylic acids :
![]()
Ex. 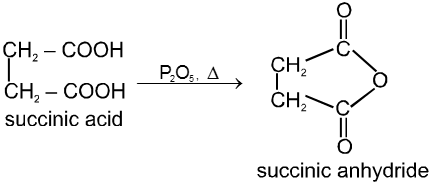
Ex.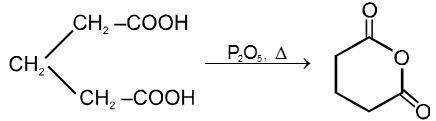
Ex. 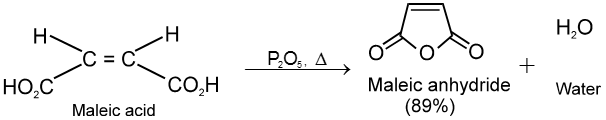
(2) From acid and acid halide :
Ex. ![]()
Ex. ![]()
Chemical Reactions
(1) Reaction with aromatic compounds (Friedel crafts acylation) :
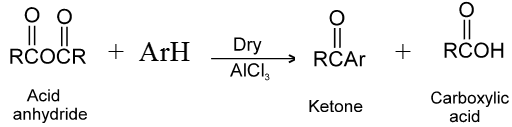
Ex.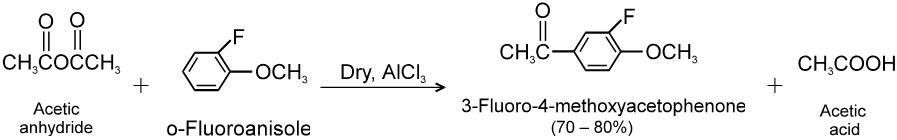
(2) Reaction with alcohols :
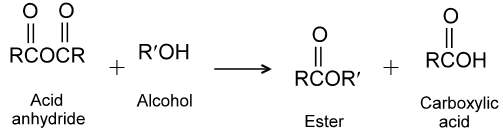
Ex.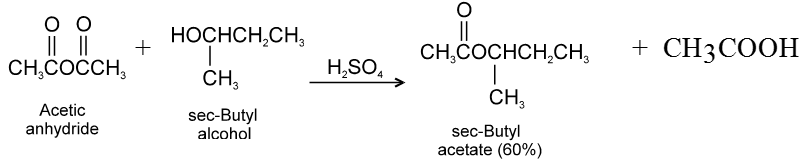
(3) Reaction with ammonia and amines :
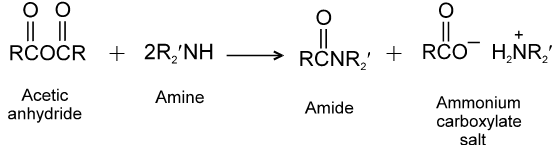
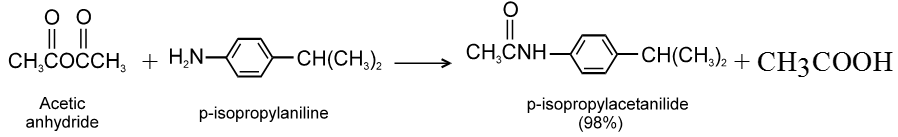
(4) Hydrolysis :
Acid anhydrides react with water to yield two carboxylic acid functions. Cyclic anhydrides yield dicarboxylic acids.
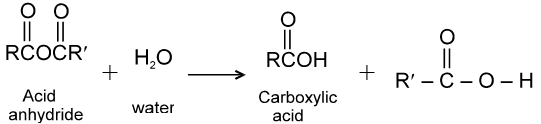
Heating Effects :
(a) Heating effect on monocarboxylic acid :
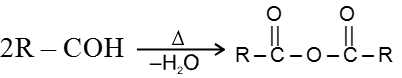
(b) Heating effect on dicarboxylic acid :
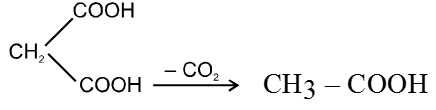
(c) Heating effects on Hydroxy acids :
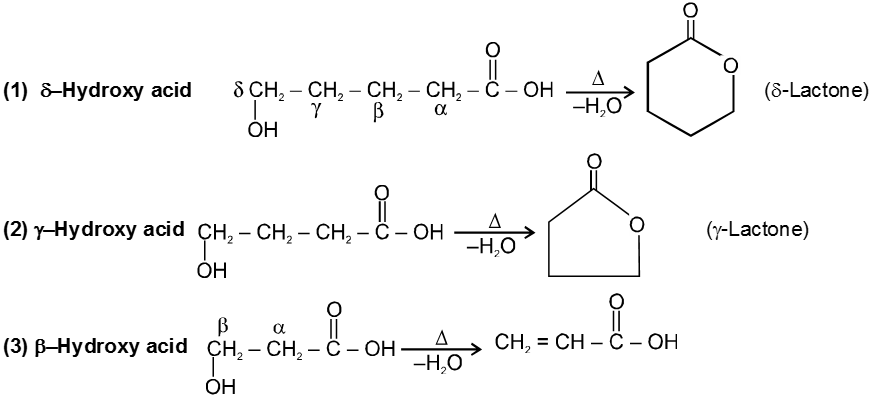
Since 4 or 8 membered rings are less stable the refore b-Hydroxy acids on heating produce a, b unsaturated carboxylic acid.
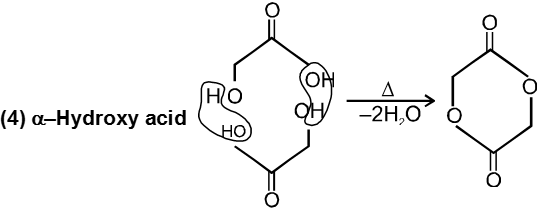
(d) Heating effects on esters :
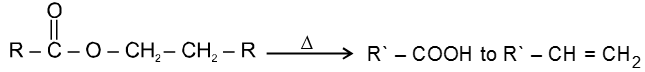
Mech :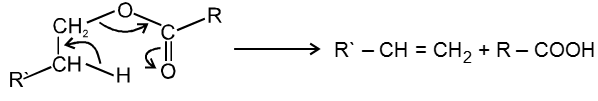
This reaction follows syn elimination & hoffman product is formed.
7. Lipids
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Lipids
Lipids are constituents of plants and tissues which are insoluble in water but soluble in organic solvents such as chloroform, carbon tetrachloride, ether or benzene. They include large variety of compounds of varying structures such oils and fats; phospholipids, steroids, etc.
Lipids are mainly made up of carbon, hydrogen and oxygen. The number of oxygen atoms in a lipid molecule is always small as compared to the number of carbon atoms. Sometimes small amounts of phophsorus, nitrogen and sulphur are also present. They have a major portion of their structure like a hydrocarbon (aliphatic 663 or fused carbon rings).
Lipids serve as energy reserve for use in metabolism and as a major structural material in cell membranes for regulating the activities of cell and tissues.
Simple lipids are esters of glycerol with highly fatty acids (saturated or unsaturated). These are generally called glycerides of fats and oils.
Waxes are esters of fatty acids with certain alcohols, not glycerol. Fats and oils have biological importance, but waxes have no value as these are not digested.
Functions of triglycerides :
(i) They are energy reserves in the cells and tissues of living system. When digested, triglycerides are hydrolyzed to fatty acids and glycerol.
(ii) Catabolism of fatty acids form acetyl-coenzyme-A . Most of the energy of fatty acids is converted into ATP.
(iii) Acetyl co-enzyme is the starting material for the synthesis of many compounds.
(iv) Fats deposited beneath the skin and around the internal organs minimise loss of body heat and also act as cushions to absorb mechanical impacts.
Another very important class of lipids are the phospholipids. These are polar lipids and like the fats, are esters of glycerol. In this case, however, only two fatty acid molecules are-esterified to glycerol, at the first and second carbon atom. The remaining end position of the glycerol is esterified to a molecule of phosphoric acid, which in turn is also esterified to another alcohol. This gives a general structure.
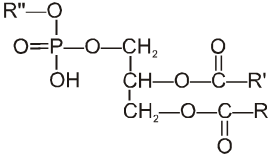
The alcoholic compound linked to phosphoric group may be ethanol, amine, serine or inositol. The phosphate group forms a polar end, i.e., hydrophilic (water-attracting) and the two fatty acid chains constitute ‘the non-polar tail, i.e., hydrophobic (water-repelling). This structure gives the phospholipids good emulsifying and membrane forming properties and hence they are used as good surfactants. Cell membranes are composed of phospholipids and proteins in about equal proportion.
The phospholipids in the membrane appear to be arranged in a double layer or bilayer in which the non-polar tails face each other, thereby exposing the polar heads to the aqueous environment on either side of the membrane. Proteins found in the membrane are embedded in the mossaic formed by the lipids.
Phospholipids facilitate the transport of ions and molecules in and out of the cell and regulate the concentration of molecules and ions within the cell. They provide structural support for certain proteins. The common examples of phospholipids are lecithins and cephanlins which are found in brain, nerve cells and liver of animals. These are also found in egg yolks, yeast, soyabeans and other foods.
Lecithins are derivatives of HOCH2CH2N(CH3)CI– and cephanlins ethanolamine, HOCH2CH2NH2•
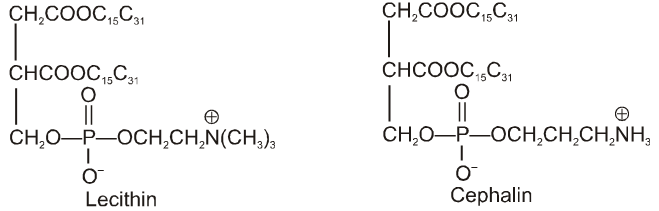
Lecithin contains a quaternary nitrogen whereas cephalin contains only primary nitrogen. The above mentioned lipids are mainly straight chain compounds. There is a third class of lipids which are not straight chain compounds. They are called Sterols. The sterols are composed of fused hydrocarbon rings and a long hydrocarbon side chain. Cholesterol is most important compound of this class and is found in animals only. It exists either free or as ester with a fatty acid. Cholesterol is also the precursor of hormones.
Cholesterol and its esters are insoluble in water. So they are deposited in the arteries and veins if the blood cholesterol rises. This leads to high blood pressure and heart diseases. Cholesterol is a part of animal cell membrane and is used to synthesise steroid hormones, vitamin-D and bile salts.
Waxes :
These are simple lipids and they are esters of long chain fatty acids and monohydric alcohols. General formula of waxes can be given as RCOOR’.
8. Enzymes and Hormones
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Enzymes and Hormones
Enzymes :
Nitrogenous complex compounds with basic nature are called Enzymes. Enzymes are biological catalysts. All biological reactions are catalysed by enzyme. Enzyme lower the activation energy thereby increasing the rate of the reaction. For example activation energy for acid hydrolysis of sucrose is 6.22 KJ mol–1, while hydrolysis by SUCRASE enzyme the activation energy is 2.15 KJ mol–1.
Composition of enzymes :
Chemically all enzymes are globular proteins. However some enzymes are also associated with some non-protein component called the cofactor. For their activity. These cofactors are of two types : -
(a) Inorganic ions, such as Zn2+, Mg2+ , Mn2+ , Fe2+ , Cu2+ , Co2+ , Mo3+ , K+, Na+ etc
(b) organic molecules :- These are also of two types.
(i) Coenzymes : These are small organic molecules which are loosely held to protein and can be easily separated by dialysis. These are usually derived from vitamins such as thiamine, riboflavin, niacin etc.
(ii) Prosthetic group : These are also organic molecules which are tightly held to protein by covalent bonds but can be separated by only careful hydrolysis. Most of these are derived from vitamins such as biotin. Enzymes are vital for biological process without them the life process would be very slow. For example with out enzyme the meal would take 50 years to digest.
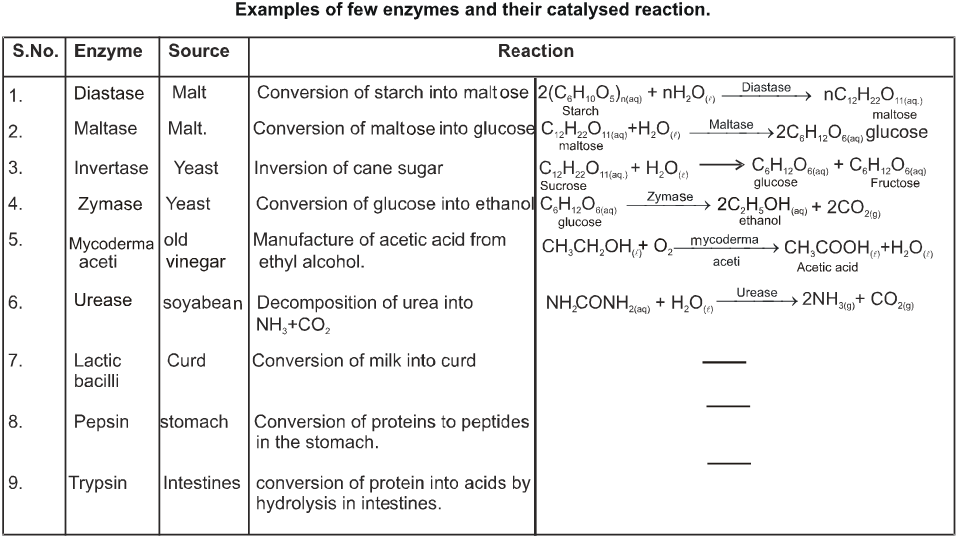
Characteristics of Enzymes :
(i) Specificity : Each enzyme catalyses only one chemical reaction.
(ii) Efficiency : Enzymes are very efficient catalyst. They increase the rate of reaction by 10 times.
(iii) Small quantity :- Only small amount of enzyme can be highly efficient because, they are regenerated after their catalytic activity
(iv) Optimum temp. and pH :- Enzymes catalyse reactions at pH of 7.4 and temperature range is 298-310 K (25ºC to 37ºC) (Exception pepsin work at pH 2–3)
Application of Enzymes :
(i) In the manufacture of BEER and WINE and vinegar by the fermentation of carbohydrates.
(ii) In the manufacture of sweet syrup from corn starch.
(iii) In the production of cheese by coagulation of milk.
(iv) Enzymes are helpfull mainly in the digestion process. Besides enzymes are used for controlling diseases such as
- Albinism diseases caused by the deficiency of the enzyme tryosinase
- streptokinase is used to dissolve blood clots in the treatment of heart disease.
Hormones :
These are the chemicals which are secreted by ductless glands and transported to different parts of the body by the blood stream where they control different physiological action of the body. The word hormone is derived from Greek language, which means to gear up or to excite. Hormones control cell and tissue growth, heart beat, blood pressure, secretion of digestive enzymes, kidney function, the reproductive system and lactation, etc. In mammals, the secretion of the hormones is controlled by anterior lobe of the pitutary gland located at the base of brain. These hormones are then carried to other gland such as adrenal cortex and sex glands to stimulate the production of other hormones.
Classification of hormones: On the basis of structure and composition, hormones are classified into following types:
(i) Steroid hormones:
These hormones are composed to steroid nucleus having following structure:
These are mostly secreted by Testes and adrenal cortex of males and hence, called as adrenal cortex hormones or sex hormones. Examples of these hormones are androgens Testosterones and dihydro testosterone. These hormones are stimulant of male sex characteristics. In females, sex hormones are estrogens. These hormones are produced in ovaries.
(ii) Polypeptides hormones :
These hormones are polypeptide (protein). These hormones are secreted by posterior lobe of the pitutary gland. eg. oxytocin, vasopressin.
(iii) Amine hormones :
These are water soluble hormones containing amino groups. These are derived from amino acids.
e.g thyroxine and adrenaline.
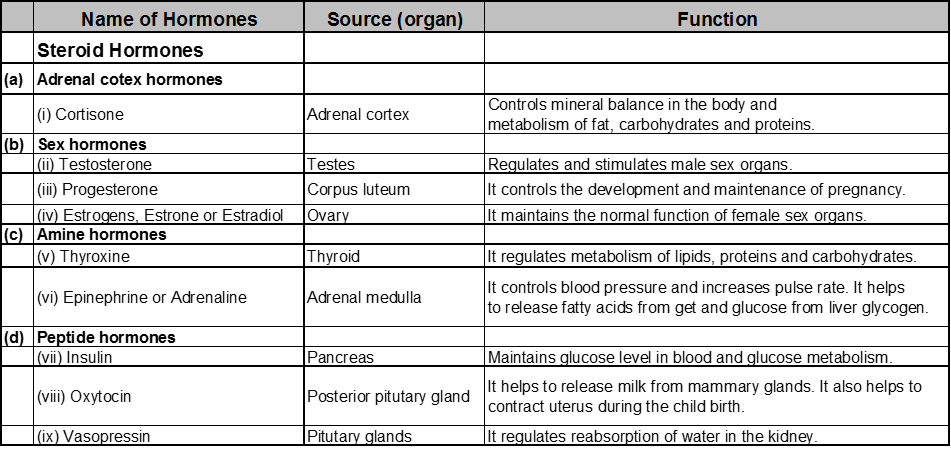
9. Vitamins
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Vitamins
Definition : Vitamins may be defined as a group of biomolecules which are required in small amounts for normal metabolic process and for the life, growth and health of human beings and animals.
Sources of Vitamins : Human body can synthesize vitamin eg. some members of vitamin B complex synthesized by micro organisms present in the intestinal tract. Most of the vitamins can not be synthesize by our body. Therefore, they must be supplied in the food. Plants can synthesize almost all vitamins. Vitamin D may either be supplied in the food or may be produced in the skin by the irradiation of ergosterol (a sterol present in our body) with ultraviolet light.
Classification of Vitamins : There is very little common to the various vitamins, therefore there are usually desiganated by alphabet letters A, B, C, D, E and K. Some of these are classified as sub groups eg. B1, B2, B3, B4, B6, B12 D1, D2 etc. About 25 vitamins are known to date. Vitamins are broadly classified into two catagories.
(i) Water soluble vitamins : Vitamin B-complex and vitamin C are water soluble vitamins and must be supplied regularly in diet.
(ii) Fat soluble vitamins : These are oily substances and soluble in fats These are A,D,E and K. They are stored in liver and adipose (fat storing) tissues.
(iii) Biotin (Vitamin H) : It is neither soluble in water nor in fats. Lack of particular vitamin causes a specific deficinecy disease. Some important vitamins their characteristics, sources and deficiency diseases are summarised in the following table.
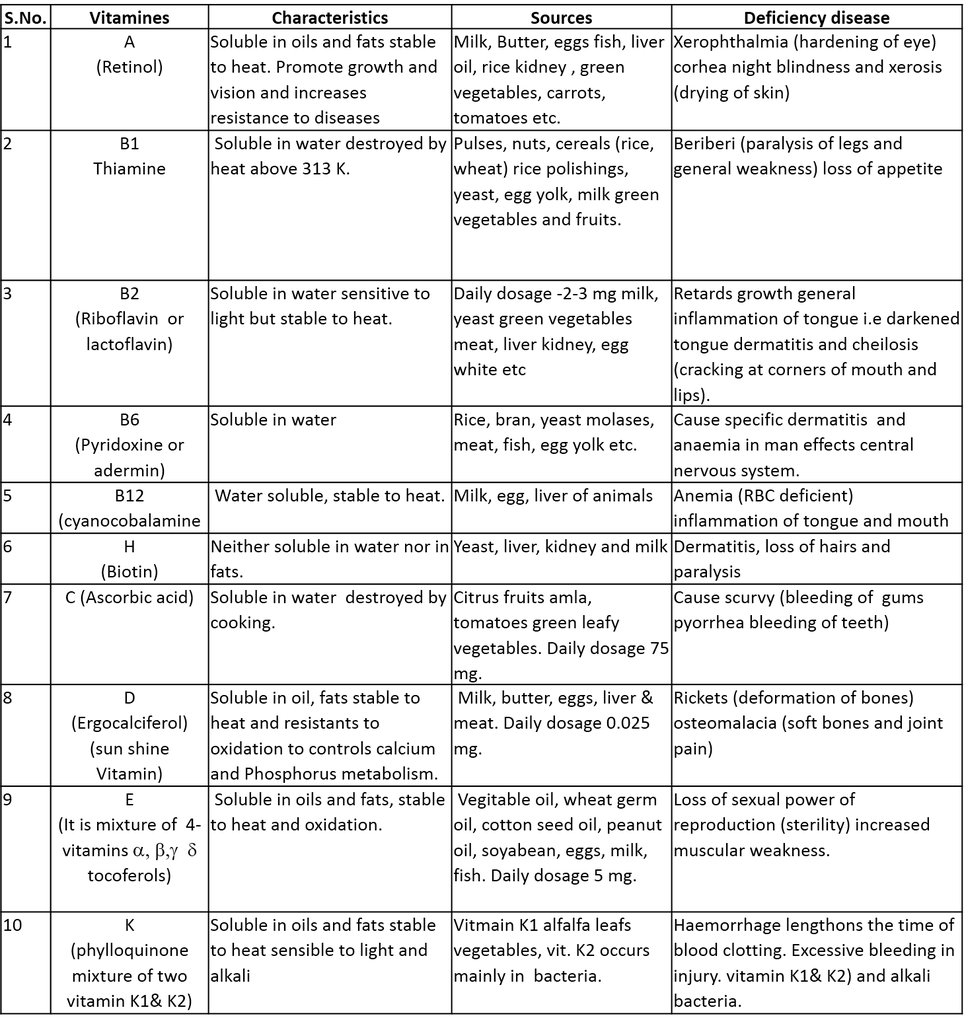
10. Physical properties of organic compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Physical properties of organic compounds
1. Bond Polarity :
A bond with electrons shared equally between the two atoms is called a nonpolar covalent bond.
Ex. Bond in H2 and the C–C bond in ethane are nonpolar covalent bonds.
Generally in most of the bonds between two different elements, the bonding electrons are attracted more strongly towards one of the two nuclei. An unequally shared pair of bonding electrons is called a polar covalent bond.
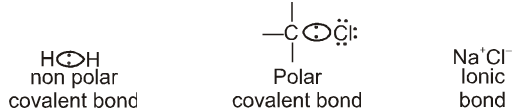
A covalent bond will be polar if atoms forming it differ in electronegativity.
Electronegativity F > O > Cl , N > Br > C,H
2. Intermolecular forces :
• Attractions between molecules are particularly important in solids and liquids. In these “Condensed” phases, the molecules are continously in contact with each other. The melting points, boiling points, and solubilities of organic compounds show the effects of these forces. Two major kinds of attractive forces cause molecules to associate into solids and liquids;
(A) Dipole-dipole interactions (B) VanderWaals forces
(A) Dipole-Dipole interaction :
Dipole-dipole interactions result from the approach of two polar molecules.
• If their positive and negative ends approach, the interaction is an attractive one.
• If two negative ends or two positive ends approach, the interaction is repulsive one.
• In a liquid or a solid the molecules are mostly oriented with the positive and negative ends together and the net forces is attractive.
Symbolized by :
![]()
• An especially strong kind of dipole- dipole attraction is hydrogen bonding
Types of Hydrogen Bonding :
(a) Intermolecular hydrogen bonding : In such type of linkages the two or more than two molecules of the same compound combine together to give a polymeric aggregate.
This phenomena is also known as association.
Example :
(I) Hydrogen bonding in carboxylic acids e.g. formic acid (Dimerisation)
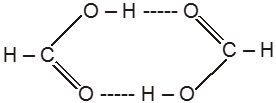
(II) In m-Chlorophenol
(III) In water
![]()
(IV) In p-Nitrophenol
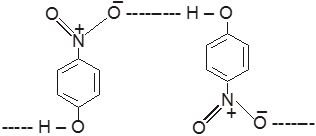
(V) In p-Nitrophenol & water
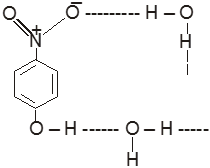
(b) Intramolecular hydrogen bonding:- In this type, hydrogen bonding occur within two atoms of the same molecule. This type of hydrogen bonding is commonly known as chelation.
Ex.
Conclusion :
(a) The chelation between the ortho substituted groups restricts the possibility of intermolecular hydrogen bonding.
(b) Chelation does not take place in m – & p – isomers because the two groups far away from each other.
3. Boiling Points :
• The boiling point (bp) of a compound is the temperature at which the compound’s vapor pressure equals the atmospheric pressure. In order for a compound to vaporize, the force that hold the molecules close to each other in the liquid must overcome. This means that the boiling point of a compound depends on the strength of the attractive forces between the individual molecules.
• If the molecules are held together by strong forces, it will take a lot of energy to pull the molecules away from each other and the compound will have a high boiling point.
• If however, the molecule are held together by weak forces, only a small amount of energy will be needed to pull the molecules away from each other and the compound will have a low boiling point.
Factors affecting boiling point :
(1) Hydrogen Bonding :
Alcohols :
Alcohols have much higher boiling points than alkanes or ethers of comparable molecular weight because, in addition to van der Waals forces and the dipole–dipole interactions of the carbon–oxygen bond alcohols can form hydrogen bonds.
The successive replacement of hydrogen atom of the –OH group of alcohol by alkyl group to form ether blocks the probability of hydrogen bonding reduces and thus B.P. of alcohols are higher than ether.
Ex. 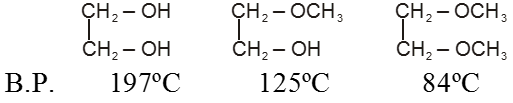
Water :
Water has the lowest molecular weight among hydrides of the VI group of periodic table, it has the highest boiling point. Water molecules associate through intermolecular hydrogen bonding and thus require more energy to separate the molecules for vaporization.
Ex. ![]()
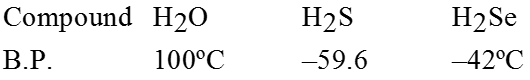
Amines :
• Primary and secondary amines also form hydrogen bonds, so these amines have higher boiling points than alkanes with similar molecular weights.
• Nitrogen is not as electronegative as oxygen, however, which means that the hydrogen bonds between amine molecules are weaker than the hydrogen bonds between alcohol molecules.
• Amines, therefore, have lower boiling points than alcohols with similar molecular weights.
• Because primary amines have two N–H bonds, hydrogen bonding is more significant for primary amines than for secondary amines. Tertiary amines cannot form hydrogen bonds with each other because they do not have a hydrogen attached to the nitrogen. Consequently if you compare amines with the same molecular weight and similar structures, primary amines have higher boiling point than secondary amines and secondary amines have higher boiling points than tertiary amines.
Ex.
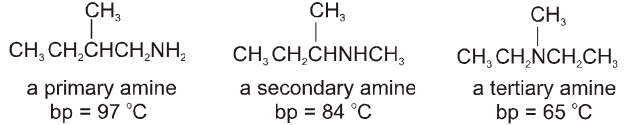
(2) Dipole - Dipole interactions :
Dipole–dipole interactions, are stronger than van der Waals forces but not as strong as ionic or covalent bonds. Ethers generally have higher boiling points than alkanes of comparable molecular weight because both van der Waals forces and dipole–dipole interactions must be overcome for an ether to boil.
Ex.

Ex. ![]()
(3) Molecular weight :
The boiling points for any homologous series of compounds increase as their molecular weights increase because of the increase in van der Waals forces. So the boiling points of a homlogous series of ethers, alkyl halides, alcohols, and amines increase with increasing molecular weight.
Ex. B. P : CH3 I > CH3Br > CH3 – Cl > CH3 – F
Ex..
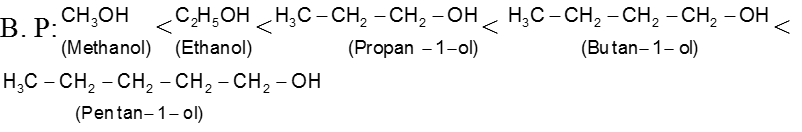
Ex.
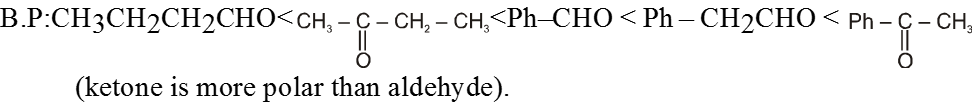
(4) Vander Waals forces
• The molecules of an alkane are held together by these induced dipole-induced dipole interactions known as van der Waals forces or London forces. In order for an alkane to boil, these van der Waals forces must be overcome.
• The homologous series of alkanes boiling points increase as their size increases. This occurs becuase each additional methylene group increases the area of contact between the molecules.
• Because the strength of van der Waals force depends on the area of contact between molecules, branching in a compound lowers its boiling point because it reduces the area of contact.
• If two alkanes have the same molecular weight, the more highly branched alkane will have a lower boiling point.
Ex.
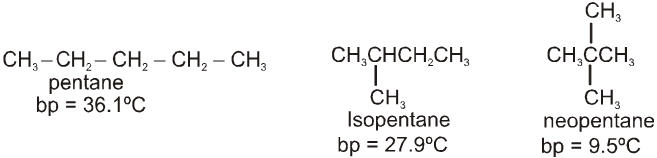
General order of boiling point of various F.G.
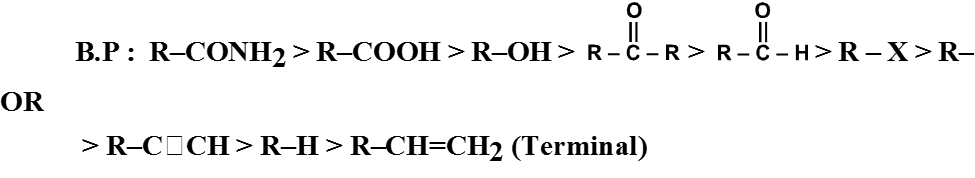
Ex. o-Chlorophenol has lower boiling point in comparison to its p-isomer.
![]() (Intramolecular H-bond in o-Chlorophenol)
(Intramolecular H-bond in o-Chlorophenol)
 (Intermolecular H-bond in p-chlorophenol)
(Intermolecular H-bond in p-chlorophenol)
4. Melting Point :
The temperature at which the thermal energy of the particles is great enough to overcome the intracrystalline forces that hold them in position is known as melting point.
• Melting is the change from the highly orderd arrangment of particles in the crystalline lattice to the more random arrangment that characterizes a liquid.
Factors affecting melting point :
(1) Molecular weight :
Melting points of alkanes increase in a homologous series as the molecular weight increases.
Ex. M.P. : CH3–CH2–CH3 < CH3CH2–CH2–CH3 < CH3–CH2–CH2–CH2–CH3
(2) Packing :
• Packing is a property that determines how well the individual molecules in a solid fit together in the crystal lattice. If molecules are more closely packed then more energy is required to break the lattice and melt the compound.
• Alkanes with an odd number of carbon atoms pack less tightly than alkanes with an even number of carbon atoms. This decreases the intermolecular forces between alkanes with odd number of carbon atoms, which decreases their melting points.
• In geometrical isomers the trans isomers are more symmetrical than cis isomers (Cab=Cab type alkenes)
so trans form have higher M.P. than cis isomers.
- The heavier the molecule and stronger the intermolecular forces, higher will be the M.P. of the compound.
- Ortho isomers of hydroxy, nitro, carbonyl compounds have low M.P. than their corresponding m – & p – isomers.
Ex.
![]()
Ex. Ortho - hydroxy, nitro-, carbonyl, carboxylic or chloro compounds have lower melting and boiling points than the respective meta or para isomer due to interamolecular H-bonding in ortho substituted compound.
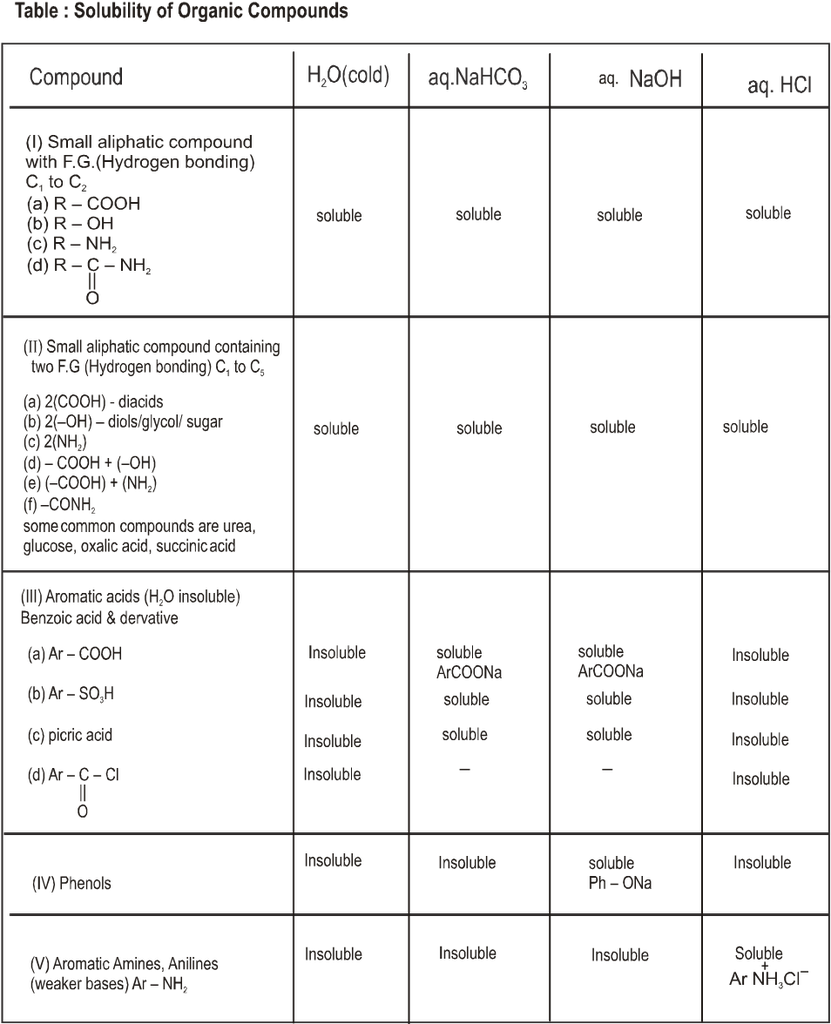
Ex. 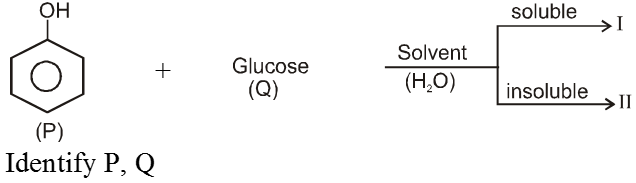
Sol. ![]()
Ex. Binary mixtures - (Two components
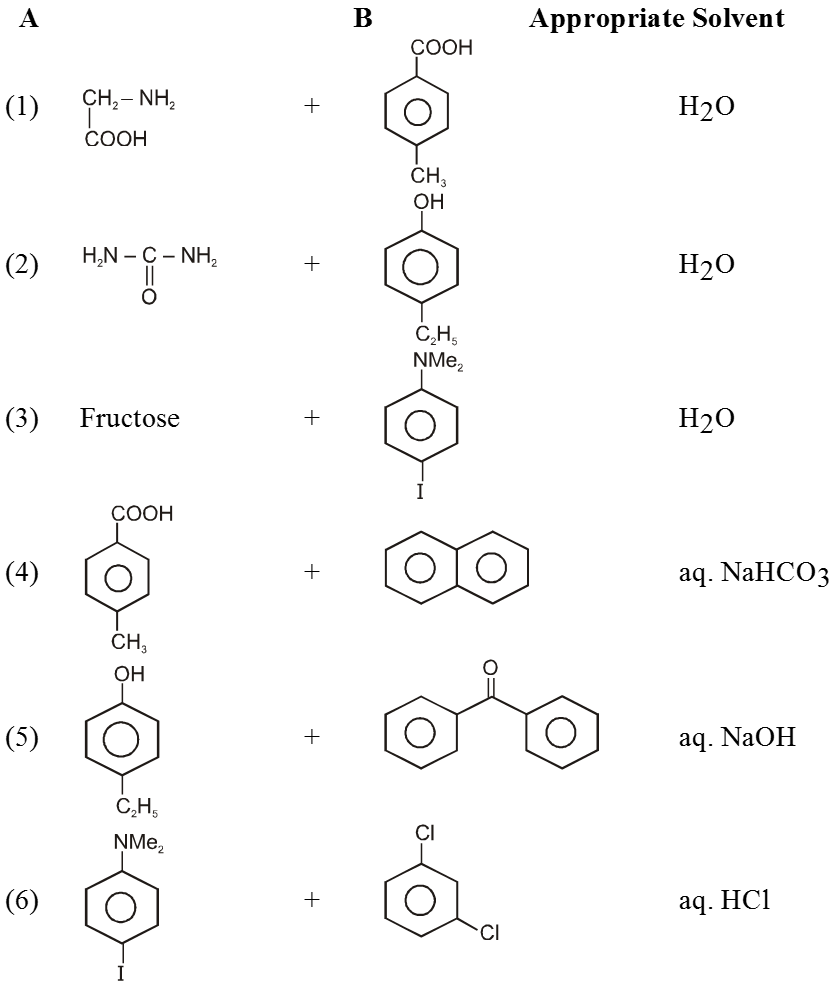
10. Nucleophilic substitution (SN) reaction of alcohols
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Nucleophilic substitution (SN) reaction of alcohols
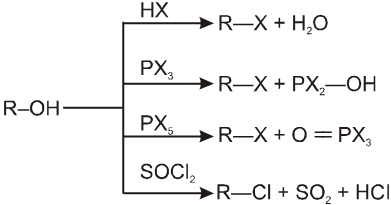
In presence of HX :
![]()
This is a nucleophilic substitution (SN1/SN2)
Mechanism :
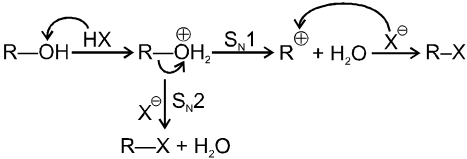
Note : Mainly b - unsubstituted 1º alcohol give SN2 reaction with HX.
Ex.
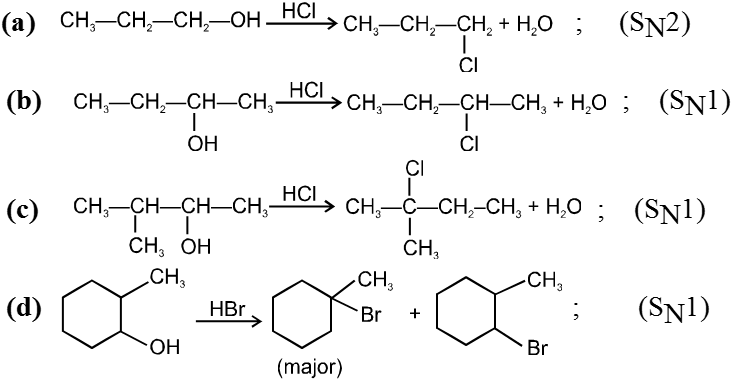
- Reactivity of HX is HI > HBr > HCl > HF.
Finkelstein reaction :
![]() (reduced) [Final product is Alkane (R-H) not R-I]
(reduced) [Final product is Alkane (R-H) not R-I]
This is a problems that is why iodides are best prepared through halogen exchange reaction. It is also known as Finkelstein reaction. In this reaction R—Cl and R—Br is treated with sodium iodide in acetone.
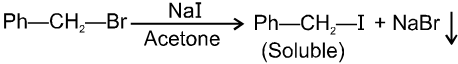
![]()
- NaI is soluble in acetone. In this reaction acetone is used because sodium iodide is soluble in acetone but NaBr and NaCl are insoluble so precipitated out. This eliminates any possibility of reverse reaction.
- It is an SN2 reaction therefore only 1ºRX and 2ºRX is used.
Lucas reagent :
The 1 : 1 mixture of anhydrous ZnCl2 : HCl (conc.) is called Lucas reagent which is used to distinguish between 1º, 2º and 3º alcohols.
![]()
Mechanism :
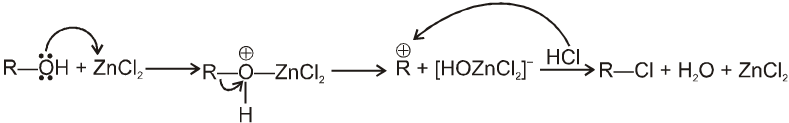
ZnCl2 increases the rate of reaction by making –OH group into a much better leaving group just through complexation.
Reactivity of alcohols is 3ºROH > 2ºROH > 1ºROH
Ex
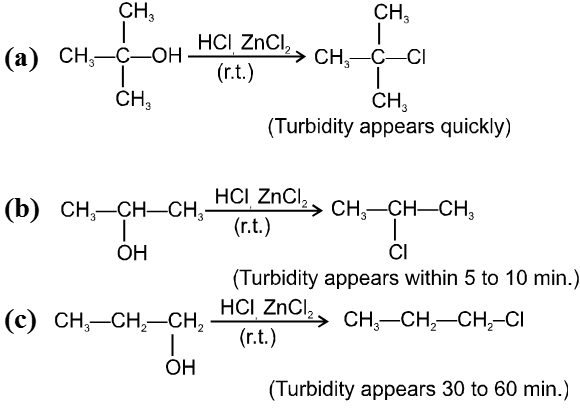
In presence of PX3 :
![]()
Mechanism :
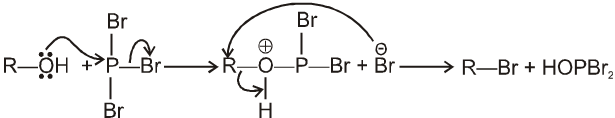
2R—OH + HOPBr2¾¾® 2R–Br +
In presence of PX5 :
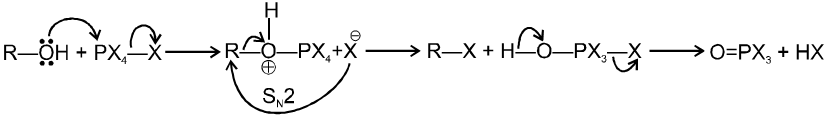
Both the PX3 & PX5 proceeds via SN2 pathways, No.rearrangement occurs.
Only 1ºR—OH and 2ºR—OH undergo this types of reaction. 3º R—OH undergoes elimination reaction.
Ex.
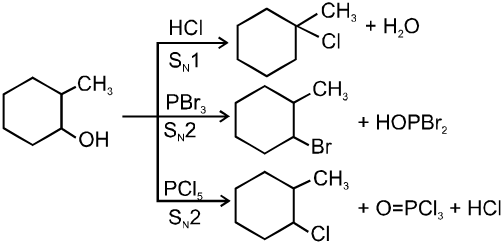
In presence of SOCl2 (Thionyl chloride) :
![]()
(A) Mechanism of SOCl2 with pyridine :
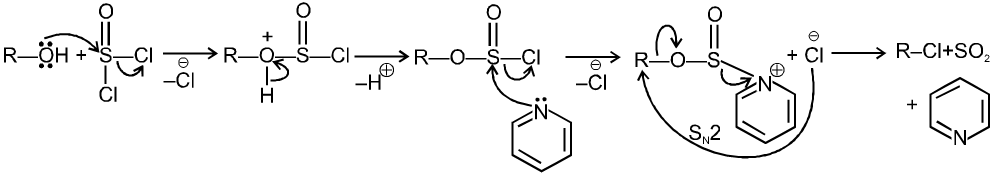
SOCl2 with pyridine undergo SN2 reaction so inversion of configuration is obtained in product.
(B) Mechanism of SOCl2 without pyridine : (Darzen method)
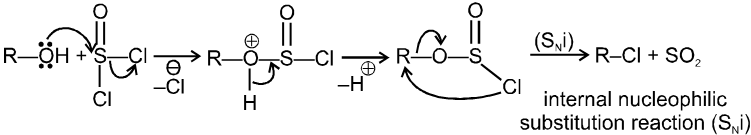
SOCl2 without pyridine or base undergo SNi reaction, so retention of configuration is obtained in product.
Ex.
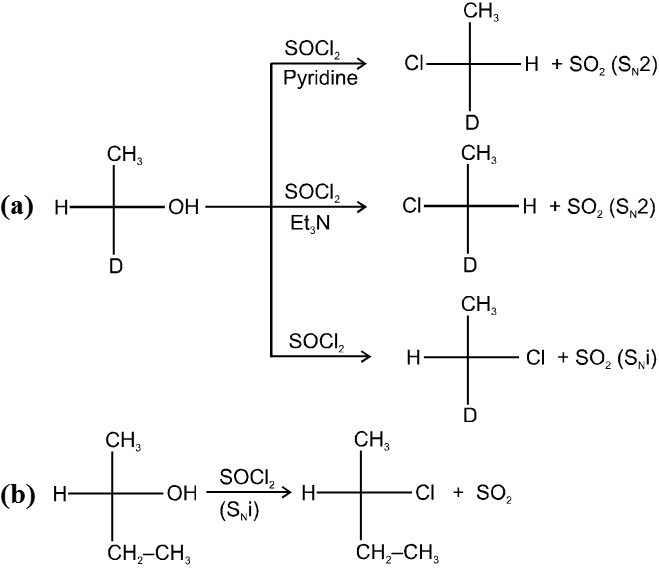
Victor Mayer test :
1° Alcohol%
![]()
2° Alcohol%
![]()
3° Alcohol %
![]()
Cerric Ammonium Nitrate test :
Alcohols(1°,2°,3°) also give characteristic red colour with cerric ammonium nitrate [(NH4)Ce(NO3)6] solution.
