 ACME SMART PUBLICATION
ACME SMART PUBLICATION
1. Coordination Entity/Coordination Sphere :
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Chapter 10:
coordination compounds
Addition Compounds :
They are formed by the combination of two or more stable compounds in stoichiometric ratio.
Double salts :
Those addition compounds which lose their identity in solution are called double salts.
K2SO4+ Al2(SO4)3 + 24H2O ® K2SO4 . Al2(SO4)3 . 24H2O![]() 2K+ (aq.) + 2Al+3 (aq.) + 4SO42– (aq.)
2K+ (aq.) + 2Al+3 (aq.) + 4SO42– (aq.)
Other examples are carnallite (KCl. MgCl2 . 6H2O), Mohr’s salt [FeSO4 . (NH4)2SO4 . 6H2O],
Potash alum [KAl(SO4)2.12H2O] etc.
Coordination Compounds :
Those addition compounds which retain their identity (i.e. doesn’t lose their identity) in solution are called coordination compounds.
Fe(CN)2 + 4KCN —® Fe(CN)2 . 4KCN or K4 [Fe(CN)6] (aq.) ![]() 4K+ (aq.) + [Fe(CN)6] 4-(aq.)
4K+ (aq.) + [Fe(CN)6] 4-(aq.)
Other examples are,
[Cu(NH3)4]SO4 (aq.) ![]() [Cu(NH3)4]2+ (aq.) + SO42– (aq.)
[Cu(NH3)4]2+ (aq.) + SO42– (aq.)
K2[Zn(CN)4] (aq.) ![]() 2K+ (aq.) + [Zn(CN)4]2– (aq.)
2K+ (aq.) + [Zn(CN)4]2– (aq.)
Coordination compound is defined as a species in which metal atom or ion is attached to group of neutral molecules / ions by coordinate covalent bonds.
Coordination Entity/Coordination Sphere :
A coordination entity constitutes a central atom/ion, usually of a metal, to which are attached a fixed number of other atoms or groups each of which is called a ligand. It may be neutral or charged. Examples being : [Co(NH3)6]3+, [PtCl4]2–, [Fe(CN)6]3–, [NiCl2(OH2)4] and (NH3), (Cl–), (CN–), (H2O) are the ligands.
The central atom/ion and the ligands attached to it are enclosed in square bracket and is collectively called as coordination sphere.
Note : The remaining ions apart from complex ions i.e. outside the coordination sphere are called counter ions, free ions or ionisable ions. For example, in K4[Fe(CN)6], the potassium (K+) ion is counter ion of coordination entity [Fe(CN)6]4–.
Central Atom/Ion :
In a coordination entity–the atom/ion to which are bound a fixed number of ligands in a definite geometrical arrangement around it, is called the central atom or ion. For example, the central atom/ion in the coordination entities : [NiCl2(OH2)4], [CoCl(NH3)5]2+ and [Fe(CN)6]3– are Ni2+, Co3+ and Fe3+, respectively. These central atoms / ions are also referred to as Lewis acids.
Ligands :
The neutral molecules, anions or cations which are directly linked with central metal atom or ion in the coordination entity are called ligands.
These may be simple ions such as Br–, small molecules such as H2O or NH3, larger molecules such as H2NCH2CH2NH2 or N(CH2CH2NH2)3 or even macromolecules such as proteins.
When a ligand is attached to a metal atom ion through a single donor atom, as with Cl–, H2O or NH3, the ligand is said to be unidentate. Similarly when a ligand is bound through two donor atoms, as in H2NCH2CH2NH2 (ethane-1, 2-diamine) or C2O42– (oxalate), the ligand is said to be bidentate and when several donor atoms are present in a single ligand as in N (CH2CH2NH2)3 or ethylenediaminetetraacetic acid (EDTA), the ligand is said to be polydentate.
Ambidentate Ligand :
Ligands which can ligate through two different atoms present in it are called ambidentate ligands. Examples of such ligands are the CN–, NO2– and SCN¯ ions. NO2– ion can coordinate through either the nitrogen or the oxygen atoms to a central metal atom/ion. Similarly, SCN¯ ion can coordinate through the sulphur or nitrogen atom. Such possibilities give rise to linkage isomerism in coordination compounds. For example,
![]() nitrito-N
nitrito-N
M ¬ O —N=O nitrito-O
M¬ SCN thiocyanato or thiocyanato-S
M¬ NCS isothiocyanato or thiocyanato-N
Chelate ligand :
Chelate ligand is a di or polydentate ligand which uses its two or more donor atoms to bind a single metal ion producing a ring. The complex formed is referred to as a chelate complex and the process of chelate formation is called chelation. The number of such ligating groups is called the denticity of the ligand.
2. Denticity and Chelation
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Denticity and Chelation :
Table : 1
Common Monodentate Ligands
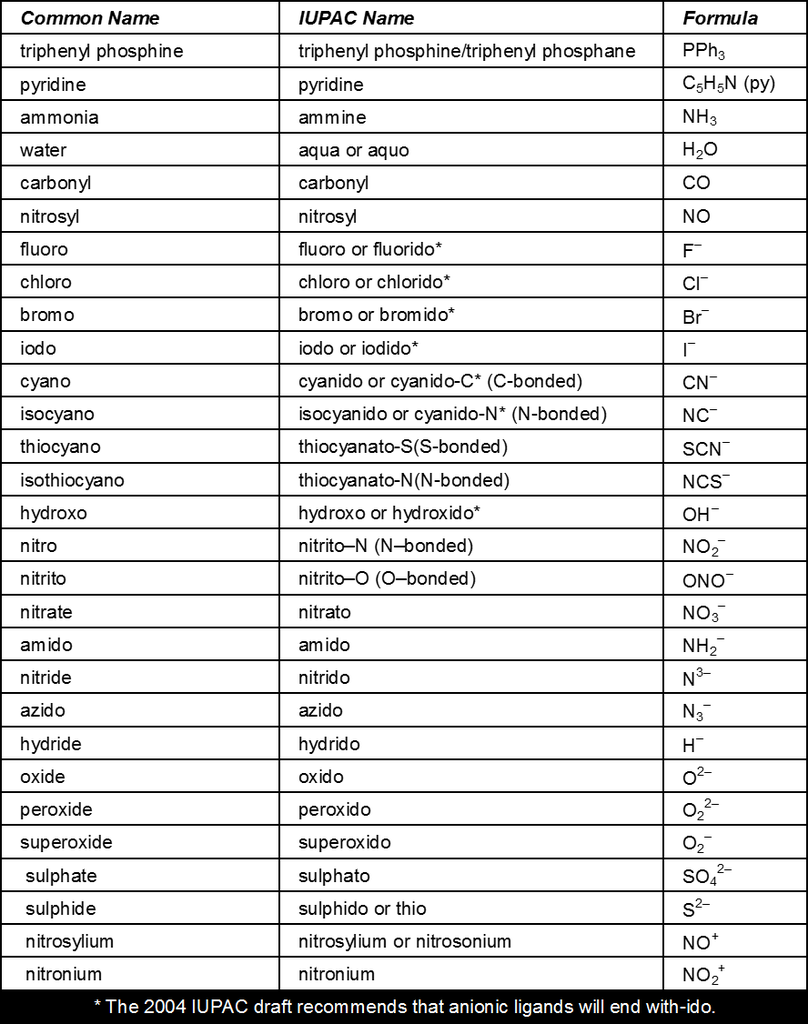
Table : 2
Common Chelating Amines

Coordination Number :
The coordination number of the central atom/ion is determined by the number of sigma bonds between the ligands and the central atom/ions i.e. the number of ligand donor atoms to which the metal is directly attached. Pi-bonds, if any, between the ligating atom and the central atom/ion are not considered for the determination of coordination number. The sigma bonding electrons may be indicated by a pair of dots, preceding the donor atom in the ligand formula as in :
[Co(NH3)6]3+, [Fe(CN)6]3–, [Ni(CO)4], [Co(Cl4)]2–.
Some common co-ordination number of important metals are as given below.

Coordination Polyhedron :
The spatial arrangement of the ligand atoms which are directly attached to the central atom/ion defines a coordination polyhedron about the central atom. Figure below shows the shapes of tetrahedral, square planar, octahedral, square pyramidal and trigonal bipyramidal coordination polyhedra. [Co(NH3)6]3+ has an octahedral geometry, while [PtCl4]2– and Ni(CO)4, are square planar and tetrahedral, respectively.
Oxidation number of Central Atom :
The oxidation number of the central atom is defined as the charge it would carry if all the ligands are removed along with the electron pairs that are shared with the central atom. Metal oxidation number is represented by a Roman numeral in parentheses following the name of the coordination entity. For example oxidation number of iron in [Fe(CN)6]3– is +3 and it is written as Fe(III).
Homoleptic and heteroleptic complexes
Complexes in which a metal is bound to only one type of donor groups, e.g., [Cr(NH3)6]3+, are known as homoleptic. Complexes in which a metal is bound to more than one type of donor groups. e.g., [Co(NH3)4Br2]+, are known as heteroleptic.
3. Nomenclature of Coordination Compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Nomenclature of Coordination Compounds
Writing the formulas of Mononuclear Coordination Entities :
The following rules are followed while writing the formulas :
(i) The central atom is placed first.
(ii) The ligands are then placed in alphabetical order. The placement of a ligand in the list does not depend on its charge.
(iii) Polydentate ligands are also placed alphabetically. In case of abbreviated ligand, the first letter of the abbreviation is used to determine the position of the ligand in the alphabetical order.
(iv) The formula for the entire coordination entity, whether charged or not, is enclosed in square brackets. When ligands are polyatomic, their formulas are enclosed in parentheses. Ligands abbreviations are also enclosed in parentheses.
(v) There should be no space between the ligands and the metal within a coordination sphere.
(vi) When the formula of a charged coordination entity is to be written without that of the counter ion, the charge is indicated outside the square brackets as a right superscript with the number before the sign. For example, [Co(H2O)6]3+, [Fe(CN)6]3– etc.
(vii) The charge of the cation(s) is balanced by the charge of the anion(s).
Writing the name of Mononuclear Coordination Compounds :
The following rules are followed when naming coordination compounds :
(i) Like simple salts the cation is named first in both positively and negatively charged coordination entities.
Examples :
[Ag(NH3)2]Cl,diamminesilver(I)chloride.K3[Fe(CN)6],potassium hexacyanidoferrate(III).
(ii) The ligands are named in an alphabetical order (according to the name of ligand, not the prefix) before the name of the central atom/ion.
Examples :
[Pt(NH3)BrCl(CH3NH2)], amminebromidochloridomethylamineplatinum(II).
[Co(H2O)2(ox)2]–, diaquabis(oxalato)cobaltate(III).
(iii) Names of the anionic ligands end in –o and those of neutral ligands are the same except aqua for H2O, ammine for NH3, carbonyl for CO, thiocarbonyl for CS and nitrosyl for NO. But names of cationic ligands end in–ium. The neutral an cationic are placed within enclosing marks ( ) .
Some more important examples of neutral and cationic ligands are :
tetraphosphorus — P4
dioxygen — O2
octasulphur — S8
(iv) Prefixes mono, di, tri, etc., are used to indicate the number of the one kind of ligands in the coordination entity. When the names of the ligands include a numerical prefix are complicated or whenever the use of normal prefixes creates some confusion, it is set off in parentheses and the second set of prefixes is used.

Example ;
[CoCl2(NH2CH2CH2NH2)2]+, dichloridobis(ethane-1,2-diamine)cobalt(III).
[NiCl2(PPh3)2], dichloridobis(triphenylphosphine)nickel(II).
(v) Oxidation state of the metal in cation, anion or neutral coordination entity is indicated by Roman numeral in the parentheses after the name of metal.
(vi) If the complex ion is a cation, the metal is named same as the element. For example, Co in a complex cation is called cobalt and Pt is called platinum. If the complex ion is an anion, the name of the metal ends with the suffix - ate. For example, Co in a complex anion, [Co(SCN)4]2– is called cobaltate. For some metals, the Latin names are used in the complex anions.

Examples ;
[Co(NH3)4Cl2]+, pentaamminechloridocobalt(III).
(NH4)2 [Co(SCN)4], ammonium tetrathiocyanato-S-cobaltate(II).
(vii) The neutral complex molecule is named similar to that of the complex cation.
Example ;
[CrCl3(py)3], trichloridotris(pyridine)chromium(III).
(viii) The prefixes cis- and trans- designate adjacent and opposite geometric locations. For examples,
[Pt(NH3)2Cl2], cis- and trans-diamminedichloridoplatinum(II),
[CoCl2(NH3)4]+, cis- and trans-tetraamminedichloridocobalt(III).
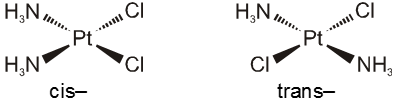
Effective Atomic Number Rule given by Sidgwick :
Effective Atomic Number (EAN) = No. of electron present on the metal atom/ion + No. of electrons donated by ligands to it.
OR
Effective Atomic Number (EAN) = Atomic no. of central metal – Oxidation state of central metal + No. of electrons donated by ligands.
The complexes in which the EAN of the central atom equals the atomic number of the next noble gas, are found to be extra stable.
Note : The EAN rule is generally found to be not valid in case of most of the complexes but in case of metal carbonyls this rule is found to be valid in all cases except one or two exceptions.
4. Werner's Theory
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Werner's Theory :
Werner in 1983 presented a theory known as Werner's coordination theory. More important postulates of this theory are :
Most element exhibit two types of valencies :
(a) Primary valency and
(b) Secondary valency.
(a) Primary valency :
This corresponds to oxidation state of the metal ion. This is also called principal, ionisable or ionic valency. It is satisfied by negative ions and its attachment with the central metal ion is shown by dotted lines.
(b) Secondary or auxiliary valency :
It is also termed as coordination number (usually abbreviated as CN) of the central metal ion. It is non-ionic or non-ionisable (i.e. coordinate covalent bond type). This is satisfied by either negative ions or neutral molecules having lone pair of electrons (e.g., H2O, NH3 etc.) or even sometimes by some positive groups. The ligands which satisfy the coordination number are directly attached to the metal atom or ion and shown by thick lines.
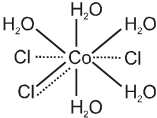
Every element tends to satisfy both its primary and secondary valencies. In order to meet this requirement a negative ion may often show a dual behaviour, i.e. it may satisfy both primary and secondary valencies since in every case the fulfillment of coordination number of the central metal ion appears essential. This dual behaviour is represented by both thick and dotted lines. For example, [CoCl(H2O)5]Cl2 is represented as
The ions/groups bound by the secondary valencies have characteristic spatial arrangements corresponding to different coordination number. In the modern terminology, such spatial arrangements are called coordination polyhedra and various possibilities are
C.N. = 2 linear
C.N. = 3 Triangular
C.N. = 4 tetrahedral or square planar
C.N. = 6 octahedral.
To distinguish between the two types of valencies, Werner introduced the square brackets [ ] to enclose those atoms making up the coordination complex and which are, therefore, not ionized.
On the basis of the above postulates Werner formulated the coordination compounds, CoCl3 . 6NH3,
CoCl3 . 5NH3 and CoCl3 . 4NH3 as : [Co(NH3)6]Cl3, [Co(NH3)5Cl]Cl2 and [Co(NH3)4Cl2]Cl respectively; the species within the square brackets being the coordination entitles (complexes) and the ions outside the square brackets the counter ions. He further postulated that octahedral, square, planar and tetrahedral geometrical shapes are more common in coordination compounds of transition metals. Thus, [Co(NH3)6]3+, [CoCl(NH3)5]2+, [CoCl2(NH3)4]+ are octahedral entities, while [Ni(CO)4] and [PtCl4]2– are tetrahedral and square-planar, respectively.
5. Bonding in coordination compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Bonding in coordination compounds
Valence bond theory :
The valence bond theory, VBT, was extended to coordination compounds by Linus Pauling in 1931. The formation of a complex involves reaction between a lewis base (ligand) and a lewis acid (metal or metal ion) with the formation of a coordinate-covalent (or dative) bonds between them. The model utilizes hybridisation of (n-1) d, ns, np or ns, np, nd orbitals of metal atom or ion to yield a set of equivalent orbitals of definite geometry to account for the observed structures such as octahedral, square planar and tetrahedral, and magnetic properties of complexes. The number of unpaired electrons, measured by the magnetic moment of the compounds determines which d-orbitals are used.
These hybrid orbitals are allowed to overlap with ligand orbitals that can donate electron pairs for bonding.
Following table provides the types of hybridisation with different coordination number.

It is to be noted that the type of hybridisation of metal and shape of complex involved can be predicted conveniently, if some characteristic of the complex like magnetic nature, geometry or whether exhibits isomerism or not, etc., be known.
Coordination Number Six.
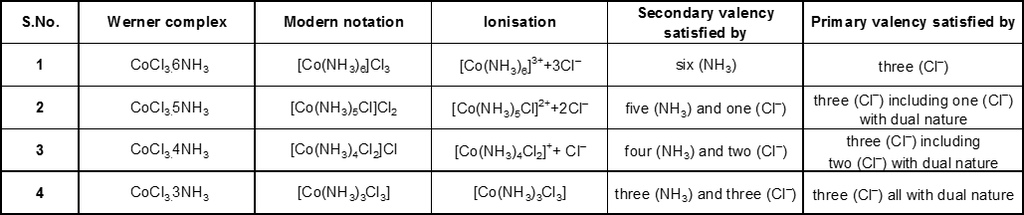
In the diamagnetic octahedral complex, [Co(NH3)6]3+, the cobalt ion is in +3 oxidation state and has the electronic configuration represented as shown below.
Co3+,[Ar]3d6 ![]()
[Co(NH3)6]3+ ![]()
![]()
Thus, the complex has octahedral geometry and is diamagnetic because of the absence of unpaired electron. Since in the formation of complex the inner d-orbital (3d) is used in hybridisation, the complex is called an inner orbital or low spin or spin paired complex.
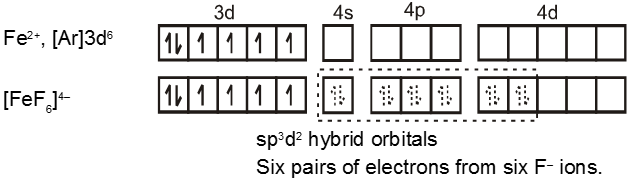
The complex [FeF6]4– is paramagnetic and uses outer orbital (4d) in hybridisation (sp3d2) ; it is thus called as outer orbital or high spin or spin free complex. So :
Coordination Number Four :
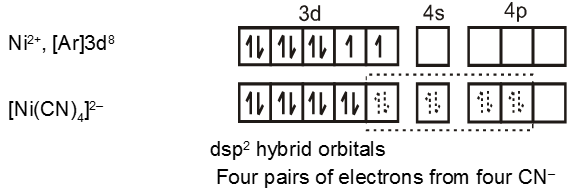
In the paramagnetic and tetrahedral complex [NiCl4]2–, the nickel is in +2 oxidation state and the ion has the electronic configuration 3d8. The hybridisation scheme is as shown in figure.
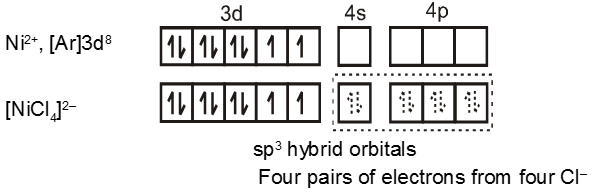
The compound is paramagnetic since it contains two unpaired electrons.
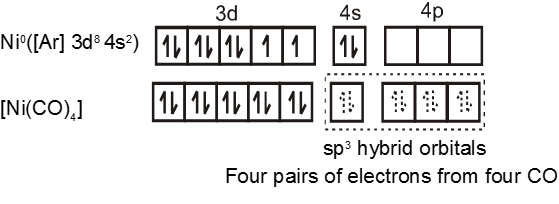
Similarly complex [Ni(CO)4] has tetrahedral geometry and is diamagnetic as it contains no unpaired electrons. The hybridisation scheme is as shown in figure.
Complexes of Pd(II) and Pt (II) are usually four-coordinate, square planar, and diamagnetic
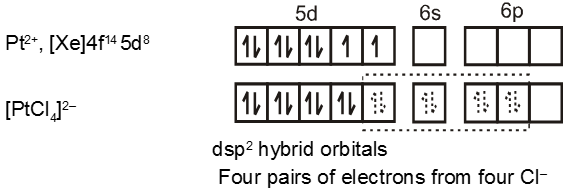
Similarly the hybridisation scheme for [Ni(CN)4]2– is as shown in figure.
While the valence bond theory, to a large extent, explains the formation, structures and magnetic behaviour of coordination compounds, it suffers from the following shortcomings :
1. A number of assumptions are involved.
2. There is no quantitative interpretation of magnetic data.
3. It has nothing to say about the spectral (colour) properties of coordination compounds.
4. It does not give a quantitative interpretation of the thermodynamic or kinetic stabilities of coordination compounds.
5. It does not make exact predictions regarding the tetrahedral and square-planar structures of 4-coordinate complexes.
6. It does not distinguish between strong and weak ligands.
6. Magnetic Properties of Coordination Compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Magnetic Properties of Coordination Compounds :
Additional information for understanding the nature of coordination entities is provided by magnetic
susceptibility measurements. We have noted that coordination compounds generally have partially filled d orbitals and as such they are expected to show characteristic magnetic properties depending upon the oxidation state, electron configuration, coordination number of the central metal and the nature of the ligand field. It is experimentally possible to determine the magnetic moments of coordination compounds which can be utilized for understanding the structures of these compounds.
The number of unpaired electrons in any complex can be easily calculated from the configuration of the metal ion, its coordination number and the nature of the ligands involved( strong or weak from the spectrochemical series) and after that the magnetic moment of the complexes can be easily calculated using
Magnetic Moment = ![]() Bohr Magneton
Bohr Magneton
For metal ions with upto three electrons in the d-orbitals like Ti3+, (d1) ; V3+ (d2) ; Cr3+ (d3) ; two vacant d-orbitals are easily available for octahedral hybridisation. The magnetic behaviour of these free ions and their coordination entities is similar. When more than three 3d electrons are present, like in Cr2+ and Mn3+ (d4) ; Mn2+ and Fe3+ (d5) ; Fe2+ and Co3+ (d6) ; the required two vacant orbitals for hybridisation is not directly available (as a consequence of Hund’s rules). Thus, for d4, d5 and d6 cases, two vacant d-orbitals are only available for hybridisation as a result of pairing of 3d electrons which leaves two, one and zero unpaired electrons respectively.
The magnetic data agree with maximum spin pairing in many cases, especially with coordination compounds containing d6 ion. However, there are complications with the coordination compounds / species having d4 and d5 ions.
[Mn(CN)6]3– has a magnetic moment equal to two unpaired electrons while [MnCl6]3– has a magnetic moment equal to four unpaired electrons.
Similarly [Fe(CN)6]3– has magnetic moment of a single unpaired electron while [FeF6]3– has a magnetic moment of five unpaired electrons. [CoF6]3– is paramagnetic with four unpaired electrons while [Co(C2O4)]3– is diamagnetic.
This anomalous behaviour is explained by valence bond theory in terms of formation of inner orbitals and outer orbitals complexes.
[Mn(CN)6]3– , [Fe(CN)6]3– and [Co(C2O4)2]3– are inner orbital complexes involving d2sp3 hybridisation, the former two are paramagnetic and the latter diamagnetic. [MnCl6]3– , [FeF6]3– and [CoF6]3– are outer orbital complexes involving sp3d2 hybridisation and are paramagnetic having four, five and four electrons respectively.
7. Crystal Field Theory
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Crystal Field Theory :
The drawbacks of VBT of coordination compounds are, to a considerable extent, removed by the Crystal Field Theory.
The crystal field theory (CFT) is an electrostatic model which considers the metal-ligand bond to be ionic arising purely from electrostatic interaction between the metal ion and the ligand. Ligands are treated as point charges in case of anions or dipoles in case of neutral molecules. The five d orbitals is an isolated gaseous metal atom/ion have same energy, i.e., they are degenerate. This degeneracy is maintained if a spherically symmetrical field of negative charges surrounds the metal atom/ion. However, when this negative field is due to ligands (either anions or the negative ends of dipolar molecules like NH3 and H2O) in a complex, it becomes asymmetrical and the degeneracy of the d orbitals is lost. It results in splitting of the d orbitals. The pattern of spitting depends upon the nature of the crystals field.
(a) Crystal field splitting in octahedral coordination entities :
In an octahedral coordination entity with six ligands surrounding the metal atom/ion, there will be repulsion between the electrons in metal d orbitals and the electrons (or negative charges) of the ligands. Such a repulsion is more when the metal d orbitals is directed towards the ligand than when it is away from the ligand. Thus, the ![]() and
and ![]() orbitals (axial orbitals) which point towards the axis along the direction of the ligand will experience more repulsion and will be raised in energy ; and the dxy , dyz and dzx orbitals (non-axial) orbitals which are directed between the axis will be lowered in energy relative to the average energy in the spherical crystal field. Thus, the degeneracy of the d orbitals has been removed due to ligand electron-metal electron repulsions in the octahedral complex to yield three orbitals of lower energy, t2g set and two orbitals of higher energy, eg set. This splitting of the degenerate levels due to the presence of ligands in a definite geometry is termed as crystal field splitting and the energy separation is denoted by D0 (the subscript o is for octahedral). Thus, the energy of the two eg orbitals will increase by (3/5)D0 and that of the three t2g will decrease by (2/5) D0.
orbitals (axial orbitals) which point towards the axis along the direction of the ligand will experience more repulsion and will be raised in energy ; and the dxy , dyz and dzx orbitals (non-axial) orbitals which are directed between the axis will be lowered in energy relative to the average energy in the spherical crystal field. Thus, the degeneracy of the d orbitals has been removed due to ligand electron-metal electron repulsions in the octahedral complex to yield three orbitals of lower energy, t2g set and two orbitals of higher energy, eg set. This splitting of the degenerate levels due to the presence of ligands in a definite geometry is termed as crystal field splitting and the energy separation is denoted by D0 (the subscript o is for octahedral). Thus, the energy of the two eg orbitals will increase by (3/5)D0 and that of the three t2g will decrease by (2/5) D0.
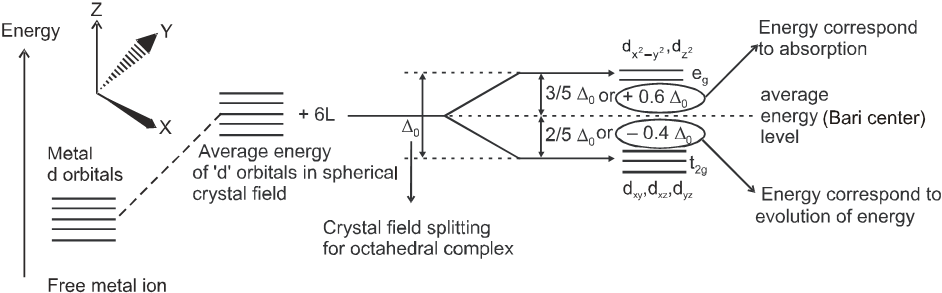
The crystal field splitting, D0, depends upon the fields produced by the ligand and charge on the metal ion. Some ligands are able to produces strong fields in which case, the splitting will be large whereas others produce weak fields and consequently result in small splitting of d orbitals. In general, ligands can be arranged in a series in the orders of increasing field strength as given below :
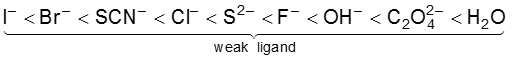 <
< 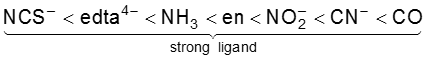
Such a series is termed as spectrochemical series. It is an experimentally determined series based on the absorption of light by complexes with different ligands. For d4 configuration, the fourth electron will singly occupy eg orbital (according to Hund’s rule) or will undergo pairing in t2g orbital, which of these possibilities occurs, depends on the relative magnitude of the crystal field splitting, D0 and the pairing energy, P (P represents the energy required for electron pairing in a single orbital). The two possibilites are :
(i) If D0 < P, the fourth electron enters one of the eg orbitals giving the configuration t32geg1. Ligands for which D0 < P are known as weak field ligands and form high spin complexes.
(ii) If D0 > P, it becomes more energetically favourable for the fourth electron to occupy a t2g orbital with configuration t2g4 eg0. Ligands which produce this effect are known as strong field ligands and form low spin complexes.
(b) Crystal field splitting in tetrahedral coordination entities :
In tetrahedral coordination entity formation, the d orbital splitting is inverted and is smaller as compared to the octahedral field splitting. For the same metal, the same ligands and metal-ligand distances, it can be shown that Dt = (4/9)D0. This may attributes to the following two reasons.
(i) there are only four ligands instead of six, so the ligand field is only two thirds the size ; as the ligand field spliting is also the two thirds the size and
(ii) the direction of the orbitals does not concide with the direction of the ligands. This reduces the crystal field spliting by roughly further two third. So Dt = ![]() ×
×![]() =
= ![]() Do.
Do.
Consequently, the orbital splitting energies are not sufficiently large for forcing pairing and, therefore, low spin configurations are rarely observed.
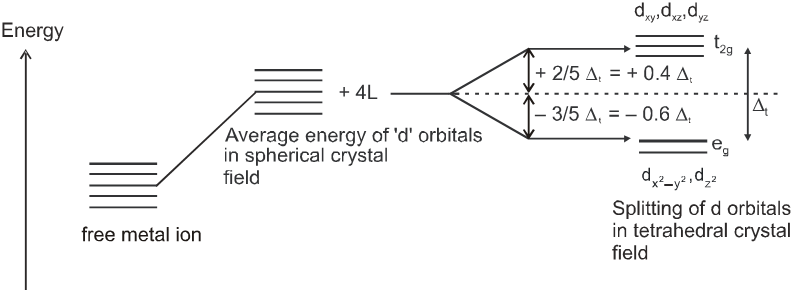
Since Dt < Do, crystal field spliting favours the formation of octahedral complexes.
8. COLOUR IN COORDINATION COMPOUNDS
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Colour in Coordination Compounds :
According to the crystal field theory the colour is due to the d-d transition of electron under the influence of ligands. The mechanism of light absorption in coordination compounds is that photons of appropriate energy can excite the coordination entity from its ground state to an excited state. Consider the Ti(III) ion in solution, that is [Ti(H2O)6]3+. This is a violet colour octahedral complex, where in the ground state of the complex a single electron is present in t2g level. The next higher state available for the transition is the empty eg level. If the light corresponding to the energy of yellow-green is absorbed by the complex, it would excite the electron from t2g level to eg level. Consequently the complex appears violet in colour.
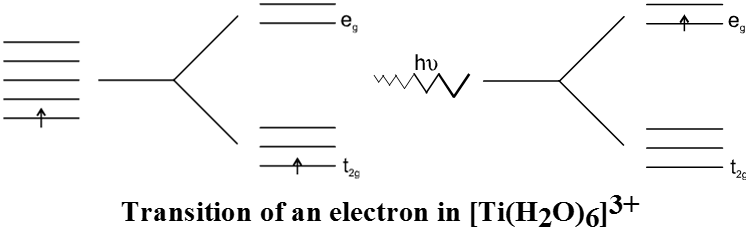
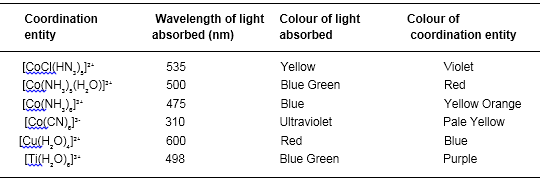
Table below gives the relationship of the wavelength absorbed and the colour observed.
Relationship between the wavelength of light absorbed and the colour observed In some coordination entitles
Note :
(a) In absence of ligand, crystal field splitting does not occur and as a consequence the substance appears colourless. For example (i) removal of water from violet coloured complex [Ti(H2O)6]Cl3 on heating makes it colourless. (ii) Similarly anhydrous copper sulphate (CuSO4) is white, but hydrated copper sulphate (CuSO4.5H2O) is blue coloured.
(b) The nature of the ligand and the molar ratio of metal : ligand also influence the colour of the complex for example in the pale green complex of [Ni(H2O)6], the colour change is absorbed when ethylenediamine is progressively added to it.
Note : Ruby is Al2O3 in which 0.5–1% Cr3+ ions (d3 electron system) are randomly distributed in the positions normally occupied by Al3+. We may consider Cr(III) species as octahedral Cr(III) complexes incorporated into the alumina lattice ; d-d transition of electron at these centres give rise to the colour (red).
Emerland is the mineral beryl (Be3Al2Si6O18) in which Cr3+ ions occupy octahedral sites, but in this case low energy corresponding to yellow red and blue is absorbed and light corresponding to green region is transmitted.
Limitations of crystal field theory
(1) It considers only the metal ion d-orbitals and gives no consideration at all to other metal orbitals (such as s, px, py and pz orbitals).
(2) It is unable to account satisfactorily for the relative strengths of ligands. For example it gives no explanation as to why H2O is a stronger ligand than OH– in the spectrochemical series.
(3) Account to this theory, the bond between the metal and ligands are purely ionic. It gives no account on the partly covalent nature of the metal ligand bonds.
(4) The CFT cannot account for the p-bonding in complexes.
9. Metal Carbonyls
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Metal Carbonyls
Compounds of metals with CO as a ligand are called metal carbonyls.
They are of two types.
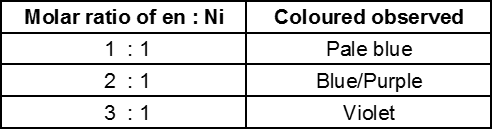
(a) Monomeric : Those metal carbonyls which contain only one metal atom per molecule are called monomeric carbonyls. For examples :[Ni(CO)4] (sp3, tetrahedral) ; [Fe(CO)5 ] (dsp3, trigonal bipyramidal)
[Cr(CO)6] (d2 sp3, octahedral) ; [V(CO)6] (d2 sp3, octahedral, only carbonyl which is paramagnetic having one unpaired electron. This is least stable among all the four carbonyls)
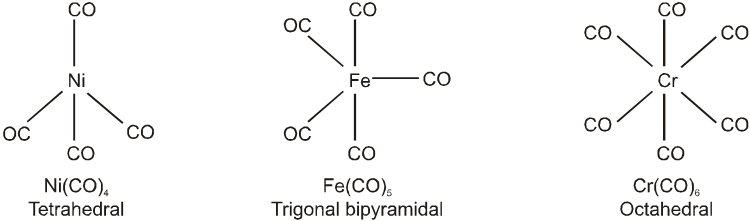
(b) Polymeric : Those metal carbonyl which contain two or more than two metal atom per molecule and they have metal-metal bonds are called polymeric carbonyl. For example : Mn2 (CO)10 , Co2(CO)8, etc.
The metal–carbon bond in metal carbonyls possess both s and p character. The M—C s bond is formed by the donation of lone pair of electrons on the carbonyl carbon (CO is a weak base) into a vacant orbital of the metal. The M — C p bond is formed by the donation of a pair of electron from a filled d orbital of metal into the vacant antibonding p* orbital of carbon monoxide. Thus carbon monoxide acts as s donor (OC ¬ M) and a p acceptor (OC ¬ M), with the two interactions creating a synergic effect which strengthens the bond between CO and the metal as shown in figure.
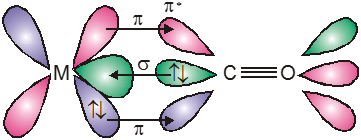
Synergic bonding
10. STABILITY OF COORDINATION COMPOUNDS
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Stability of coordination compounds :
The stability of a coordination compound [MLn] is measured in terms of the stability constant (equilibrium constant) given by the expression,
bn = [MLn]/[M(H2O)n][L]n
for the overall reaction :
M(H2O)n + nL ![]() MLn + nH2O
MLn + nH2O
By convention, the water displaced is ignored, as its concentration remains essentially constant. The above overall reaction takes place in steps, with a stability (formation) constant, K1, K2, K3, ...... Kn for each step as represented below :
M(H2O)n + L ![]() ML(H2O)n–1 + H2O
ML(H2O)n–1 + H2O
K1 = [ML(H2O)n–1] / {[M(H2O)n][L]}
MLn–1 (H2O) + L ![]() MLn + H2O
MLn + H2O
Kn = [MLn] / {[MLn–1 (H2O)] [L]}
M(H2O)n + nL![]() MLn + nH2O
MLn + nH2O
bn = K1 x K2 x K3 x ........ x Kn
bn, the stability constant, is related to thermodynamic stability when the system has reached equilibrium. Most of the measurements have been made from aqueous solutions, which implies that the complex is formed by the ligand displacing water from the aqua complex of the metal ion. Ignoring the charge and taking L as an unidentate ligand, the stepwise formation of the complex is represented as shown above. K1, K2, K3 ..... Kn representing the stepwise stability (or formation) constants.
The above is thermodynamic stability criteria, there can be another kind of stability called Kinetic Stability, which measures the rate of ligand replacement.
Some important generalisation regarding stability constants :
For a given metal and ligand the stability is generally greater when the charge on the metal ion is greater. Thus, stability of coordination entities of ions of charge 3+ is greater than the entities of 2+ ions. Further, for the divalent ions of the first row transition elements, irrespective of the ligand involved, the stabilities vary in the Irving-Williams order :
MnII < FeII < CoII < NiII < CuII > ZnII
This order is according to the size of the ions, smaller the size of the ion or greater the charge density on the metal greater is the stability of the complex.
In F¯, Cl¯, Br¯, I¯ ; F¯ forms strongest complexes due to small size & hence high charge density.
The stability also depends on the formation of chelate rings. If L is an unidentate ligand and L–L, a didentate ligand and if the donor atoms of L and L–L are the same element, then L–L will replace L. The stabilisation due to chelation is called the chelate effect. It is of great importance in biological systems and analytical chemistry. The chelate effect is maximum for the 5- and 6-membered rings. In general, rings provide greater stability to the complex.
11. Isomerism in coordination compounds :
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Isomerism in coordination compounds :
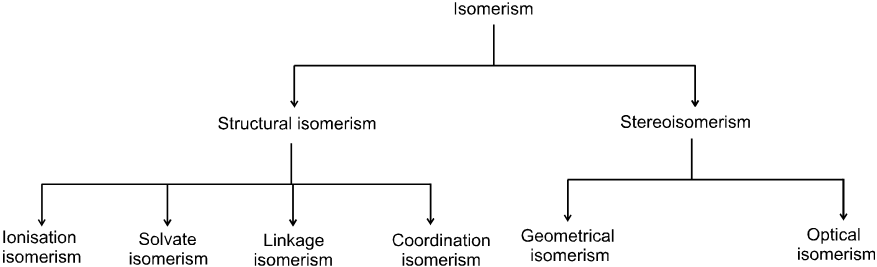
(1) Structural isomerism :
The structural isomerism arises due to different lignads within the coordination sphere.
(A) Ionisation Isomerism
This type of isomerism occurs when the counter ion in a coordination compound is itself a potential ligand and can displace a ligand which can then become the counter ion. For example, following complexes show ionisation isomerism.
[Co(NH3)5SO4]NO3 and [Co(NH3)5NO3]SO4
[Co(NH3)4(NO2)Cl]Cl and [Co(NH3)4Cl2]NO2.
[Co(NH3)4(H2O)Cl]Br2 and [Co(NH3)4BrCl]Br.H2O. [Also an example of hydrate isomers.]
[Pt(NH3)4Cl2]Br2, and [Pt(NH3)4Br2]Cl2.
[CoCl(en)2(NO2)]SCN, [Co(en)2(NO2)SCN]Cl and [Co(en)2(SCN)Cl]NO2
(B) Solvate/Hydrate Isomerism :
It occurs when water forms a part of the coordination entity or is outside it. This is similar to ionisation isomerism. Solvate isomers differ by whether or not a solvents molecule is directly bonded to the metal ion or merely present as free solvent molecules in the crystal lattice. For example, CrCl3 . 6H2O exists in three distinct isomeric forms : [Cr(H2O)6]Cl3, violet ; [CrCl(H2O)5]Cl2.H2O, blue green : [CrCl2(H2O)4]Cl.2H2O, dark green.
(C) Linkage isomerism :
In some ligands, like ambidentate ligands, there are two possible coordination sites. In such cases, linkage isomerism exist. e.g., NO2 group can be bonded to metal ions through nitrogen (–NO2) or through oxygen (–ONO). SCN too can be bonded through sulphur (–SCN) thiocyanate or through nitrogen (–NCS) isothiocyanate.
For example : [Co ONO (NH3)5] Cl2 & [Co NO2 (NH3)5] Cl2 .
(D) Coordination Isomerism
Coordination compounds made up of cationic and anionic coordination entities show this type of isomerism due to the interchange of ligands between the cation and anion entities. Some of the examples are :
(i) [Co(NH3)6][Cr(CN)6] and [Cr(NH3)6](Co(CN)6]
(ii) [Cu(NH3)4](PtCl4] and [Pt(NH3)4][CuCl4]
(iii)[Co(NH3)6][Cr(SCN)6]and[Cr(NH3)4(SCN)2](Co(NH3)2(SCN)4]
(iv) [Pt(NH3)4][PtCl6] and [Pt(NH3)4Cl2][PtCl4]
Such isomers are expected to have significant differences in their physical and chemical properties.
(2) Stereoisomerism
The isomers in which atoms are bonded to each other in the same order but that differ in the arrangement of these atoms in the space are called as stereoisomers and the phenomenon as stereoisomerism.
(A) Geometrical Isomerism
This type of isomerism arises in heteroleptic complexes due to different possible geometric arrangements of the ligands. Geometrical isomerism is common among coordination compounds with coordination numbers 4 and 6.
(i) Coordination Number Four :
(a) Tetrahedral Complex :
The tetrahedral compounds can not show geometrical isomerism as we all know that all four positions are equivalent in tetrahedral geometry.
(b) Square Planar Complex :
In a square planar complex of formula [Ma2b2] [a and b are unidentate], the two ligands ‘a’ may be arranged adjacent to each other in a cis isomer, or opposite to each other in a trans isomer as depicted.
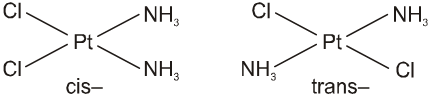
Geometrical isomers (cis and trans) of Pt(NH3)2Cl2 .
Square planar complex of the type Ma2bc (where a,b,c are unidentates) shows two geometrical isomers.

Square planar complex of the type Mabcd (where a,b,c,d are unidentates) shows three geometrical isomers.

Example is [Pt(NH3)BrCl(py)]. three isomers of the complex [Pt(NH3)(NH2OH)(py)(NO2)]+ have been isolated and identified.
Square planar complex of the type M(AB)2 (where AB are unsymmetrical bidentates) shows two geometrical isomers. Example is [Pt(gly)2] in which gly is unsymmetrical ligand.
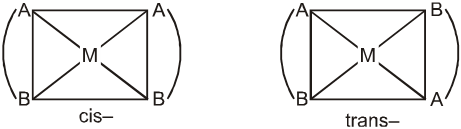
Similarly, M(AB)(CD) also shows two geometrical isomers.
Note : M(AA)2, (where AA are symmetrical bidentates) does not show geometrical isomerism. e.g., [Cu(en)2]2+ [Pt(ox)2]2–, etc.
(ii) Coordination Number Six :
Geometrical isomerism is also possible in octahedral complexes.
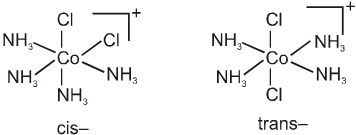
Geometrical isomers (cis and trans) of [Co(NH3)4Cl2]+
Number of possible isomers and the spatial arrangements of the ligands around the central metal ion for the specific complexes are given below.
(I) Complexes containing only unidentate ligands
(a) Ma2b4 – 2
(b) Ma4bc – 2
(c) Ma3b3
Complexes of the formula Ma3b3, where a and b are monodentate ligands, may show two isomeric forms called fac– and mer–. Facial isomers have three identical ligands on one triangular face where as meridional isomers have three identical ligands in a plane bisecting the molecule. Similar isomers are possible with some chelating ligands.
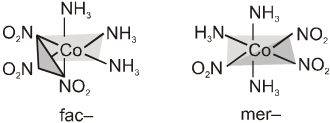
The facial(fac) and meridional(mer) isomers of [Co(NH3)3(NO2)3].
Unsymmetrical bidentate ligands also show fac-mer isomerism
(d) Ma3b2c – 3
(e) Ma3bcd – 4
(f) Ma2b2c2 – 5
(g) Ma2b2cd – 6
(h) Ma2bcde – 9
(i) Mabcdef, [Pt(py)(NH3)(NO2)(Cl)(Br)(I)] – 15
(II) Compounds containing bidentate ligand and unidentate ligands.
(i) M(AA)a3b – Two geometrical isomers are possible.
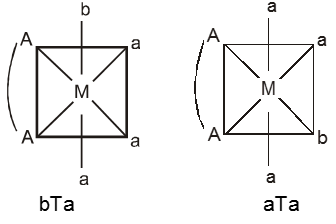
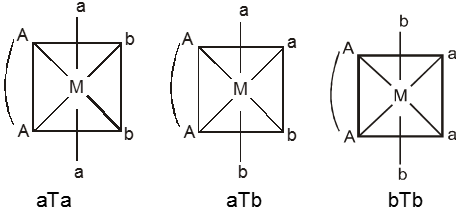
(ii) M(AA)a2b2 – Three geometrical isomers are possible.
(B) Optical Isomerism :
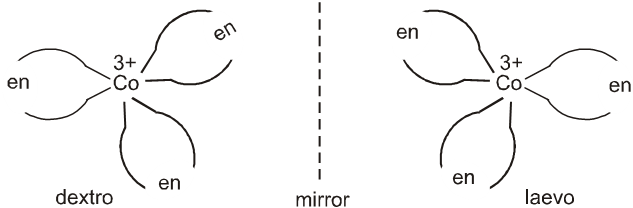
A Coordination compound which can rotate the plane of polarised light is said to be optically active. When the coordination compounds have same formula but differ in their ability to rotate directions of the plane of polarised light are said to exhibit optical isomerism and the molecules are optical isomers. Optical isomers are mirror images that cannot be superimposed on one another. These are called as enantiomers. The molecules or ions that cannot be superimposed are called chiral. This is due to the absence of elements of symmetry in the complex. The two forms are called dextro(d) and laevo(l) depending upon the direction they rotate the plane of polarised light in a polarimeter (d rotates to the right,l to the left).
(a) Octahedral complexes :
Optical isomerism is common in octahedral complexes involving didentate ligands. For example, [Co(en)3]3+ has d and l forms as given below
Cis-isomer of [PtCl2(en)2]2+ show optical isomerism as shown below because of the absence of plane of symmetry as well as centre of symmetry.
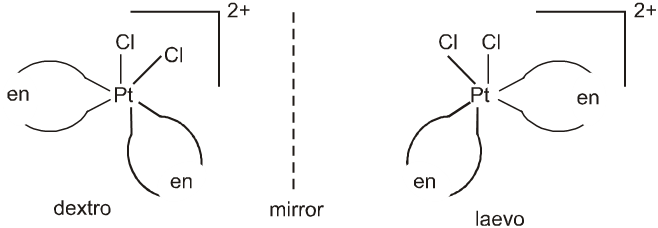
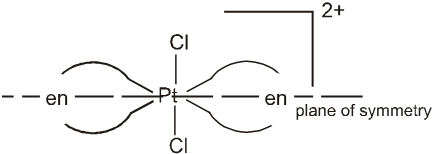
But trans isomer of [PtCl2(en2)]2+ does not show optical isomerism.
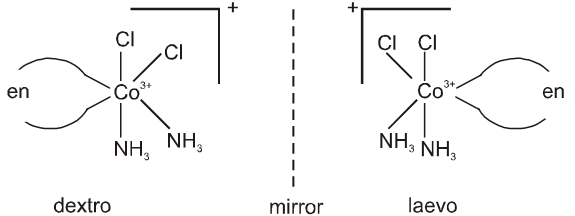
cis-[Co(NH3)2Cl2(en)]+ can show optical isomerism due to the absence of plane of symmetry as well as centre of symmetry.
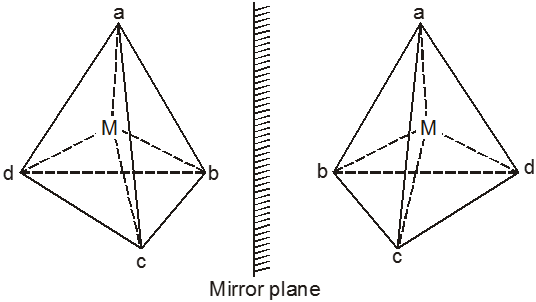
(b) Tetrahedral Complexes.
Optical isomerism is expected in tetrahedral complexes of the type [Mabcd] analogous to tetrahedral carbon atom.
(c) Square Planar Complexes.
Square planar complexes are rarely found to show the optical isomerism. The plane formed by the four ligating atoms and the metal ion is considered to be a mirror plane and thus prevents the possibility of chirality.
12. Applications of coordination and organometallic compounds
- Books Name
- ACME SMART COACHING Chemistry Book
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Chemistry
Applications of coordination and organometallic compounds
(i) Coordination compounds are of great importance in biological systems. Example being – chlorophyll (the green pigment in plants); haemoglobin (the red pigment of blood, which acts as oxygen carrier) along with myoglobin (which stores oxygen and is a regulator of respiration); Vitamin B12, cyanocobalammine, the anti-pernicions anaemia factor. All of these, respectively, are the coordination compounds of magnesium, iron and cobalt with the macrocyclic porphyrin and corrin ligands.
(ii) There are many examples of the use of coordination compounds in qualitative and quantitative chemical analysis. The familiar colour reactions given by metal ions with a number of ligands (especially the chelating ligands), as a result of formation of coordination entities, form the basis for their detection and estimation by classical and instrumental methods of analysis. Familiar examples of such reagents are : ethylenediaminetetraaceticacid (EDTA), dimethylglyoxime, a-nitroso b-naphthol, cupron, etc.
(iii) Some important extraction processes of metals, like those of extraction of silver and gold, make use of complex formation. Gold, for example, combines with cyanide in the presence of oxygen and water to form the coordination entity [Au(CN)2]– in aqueous solution. Gold can be precipitated from this solution by the addition of Zinc.
(iv) Purification of metals can be achieved through formation and subsequent decomposition of their coordination compounds. For example, impure nickel is converted to [Ni(CO)4], which is decomposed to yield pure nickel.
(v) Organometallic compounds are used as catalysts. These catalysts are either of the homogeneous type (soluble in the reaction medium) or of the heterogeneous type (insoluble in the reaction medium). The catalysed polymerisation of alkenes at atmospheric pressure and ambient temperature using Ziegler-Natta catalyst (titanium tetrachloride plus triethylaluminium) is one of the important discoveries of organometallic chemistry. The first effective homogeneous catalyst chloridotris(triphenylphosphine) rhodium(I), [RhCl(PPh3)3] for hydrogenation was given by Wilkinson.
(vi) Tetra ethyl lead (TEL) is used as antiknock compound in gasoline.
(vii) Articles can be electroplated with silver and gold much more smoothly and evenly from solutions of the complexes, [Ag(CN)2]– and [Au(CN)2]– than from a solution of simple metal ions.
(viii) In black and white photography, the developed film is fixed by washing with hypo solution which dissolves the undecomposed AgBr to form a complex ion, [Ag(S2O3)2]3–.
(ix) The chelate therapy is now a day is used in medicinal chemistry.
(a) Excess of copper and iron are removed by the chelating ligands D-penicillamine and desferrioxime B via the formation of coordination compound in plant/animal systems.
(b) EDTA is used in the treatment of lead poisoning.
(c) Coordination compounds of platinum like cis-platin and related compounds effectively inhibit the growth of tumours.
