 ACME SMART PUBLICATION
ACME SMART PUBLICATION
principle of biotechnology
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
PRINCIPLES OF BIOTECHNOLOGY
Biotechnology can be broadly defined as "using living organisms or their products for commercial purposes".
As such, biotechnology has been practiced by human society since the beginning of recorded history in such activities as baking bread, brewing alcoholic beverages, or breeding food crops or domestic animals.
A narrower and more specific definition of biotechnology is "the commercial application of living organisms or their products, which involves the deliberate manipulation of their DNA molecules".
This definition implies a set of laboratory techniques developed within the last 20 years that have been responsible for the tremendous scientific and commercial interest in biotechnology.
Some other available definitions of biotechnology are :
''The application of biological organism, system or processes to manufacturing & service industries".
-British Biotechnologist
"The integrated use of biochemistry, microbiology & genetic engineering sciences in order to achieve technological (industrial) application of capabilities of micro-organisms, cultured tissue cells & parts there of".
-European Federation of Biotechnology
"The controlled use of biological agents, such as micro-organisms or cellular components, for beneficial use ".
-US National Science Foundation
Development of biotechnology may be studied considering its growth that occurred in two phases : (1) The traditional (old) biotechnology, and (2) The new (modern) biotechnology.
1. Traditional Biotechnology:
It includes the processes that are based on the natural capabilities of microorganisms.
The traditional biotechnology is also called conventional technology which has been used for many centuries.
Curd, vinegar, ghee, wine and beer and other alcoholic beverages, idli, dosa, cheese, paneer and some other foods have been produced using traditional biotechnology.
In Indian Ayurveda, production of 'Asva', 'Arista', etc. is done through traditional biotechnology.
According to some people, traditional biotechnology, is therefore, called an art rather than a science.
2. Modern Biotechnology:
When highly new and useful traits in crop varieties and animal breeds are created with the help of genetic engineering, it is called modern biotechnology.
It was developed during 1970.
For example, in vitro fertilization leading to a 'test tube baby', synthesizing a gene and using it, developing a DNA vaccine or correcting a defective gene are all a part of modern biotechnology.
Among many, the two main techniques that gave birth to modern biotechnology are as follows:
(i) Genetic Engineering: The techniques which change the chemistry of genetic material (DNA and RNA) to introduce these into host organisms and thus alter the phenotype of the host organism are called genetic engineering (= "Recombinant DNA Technology").
(ii) To maintain the microbial contamination free (Sterile) surrounding in chemical engineering: Due to such type of maintenance only desired microorganisms/cells will be formed in large number for manufacture of biotechnological products such as antibiotics, vaccines, enzymes, hormones, blood clotting factors, etc. It is essential to have complete aseptic conditions.
Concept of Genetic Engineering
Combining DNA from different existing organisms like plants, animals, bacteria etc. results in modified organism with a combination of trait from the parents.
This sharing of DNA information occurs naturally through sexual reproduction and has been exploited in plant and animal breeding for number of years.
However sexual reproduction (recombination) can occur between individuals of same species.
Genetic engineering is the manipulation of prokaryotic as well as eukaryotic DNA.
It involves breakage of a DNA molecule at two desired places to isolate a specific DNA segment and then insert it in another DNA molecule at a desired position.
The product thus obtained is called recombinant DNA and the technique often called genetic engineering.
The cutting of the DNA at specific location became possible by so-called 'molecular scissors', i.e., restriction enzymes.
In chromosomes, there is specific 'ori' site or origin of replication which initiates the replication.
Therefore, for duplication of any foreign DNA in an organism, it should be linked with the 'ori' sites so that the foreign DNA can duplicate and multiply within the organism.
This is called gene cloning and can produce multiple copies of any template DNA.
The construction of the first recombinant DNA emerged from the possibility of linking a gene encoding antibiotic resistance with a native plasmid (autonomously replicating circular extra-chromosomal DNA) of Salmonella typhimurium.
Stanley Cohen and Herbert Boyer accomplished this in 1972 by isolating the antibiotic resistance gene by cutting out a piece of DNA from a plasmid which was responsible for conferring antibiotic resistance.
These plasmid DNA molecules act as vectors to transfer the piece of DNA attached to it.
As mosquito acts as an insect vector to transfer the malarial parasite into human body.
In the same way, a plasmid can be used as vector to deliver an alien piece of DNA into the host organism.
The linking of antibiotic resistance gene with the plasmid vector became possible with the enzyme DNA ligase, which acts on cut DNA molecules and joins their ends.
This makes a new combination of circular autonomously replicating DNA created in vitro and is known as recombinant DNA.
When this DNA is transferred into Escherichia coli, a bacterium closely related to Salmonella, it could replicate using the new host's DNA polymerase enzyme and make multiple copies.
The ability to multiply copies of antibiotic resistance gene in E. coli was called cloning of antibiotic resistance gene in E. coli.
Therefore, there are three basic steps in genetically modifying an organism :
(i) Identification of DNA with desirable genes;
(ii) Introduction of the identified DNA into the host;
(iii) Maintenance of introduced DNA in the host and transfer of the DNA to its progeny .
tools of recombinant DNA technology(RDT)
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
TOOLS OF RECOMBINANT DNA TECHNOLOGY
The technology or genetic engineering involves restriction enzymes, ligase enzymes, polymerase enzymes, vectors and the host organism.
Restriction Enzymes
In the late 1960's, scientists Stewart Linn and Werner Arber isolated samples of the two types of enzymes responsible for phage growth restriction in Escherichia coli (E. coli) bacteria.
One of these enzymes methylated DNA, while the other cleaved unmethylated DNA at a wide variety of locations along the length of the molecule. The first type of enzyme was called a "methylase" while the other was called a "restriction nuclease".
These enzymatic tools were important to scientists who were gathering the tools needed to "cut and paste" DNA molecules.
What was needed now was a tool that would cut DNA at specific sites, rather than at random sites along the length of the molecule, so that scientists could cut DNA-molecules in a predictable and reproducible way.
Site-specific Nuclease
This important development came when H.O. Smith, K.W. Wilcox, and T.J. Kelley isolated and characterized the first restriction nuclease whose functioning depended on a specific DNA nucleotide sequence.
Working, with Haemophilus influenzae bacteria, this group isolated an enzyme, called Hind II, that always cut DNA molecules at a particular point within a specific sequence of six base pair.
This sequence is :
5' G T (pyrimidine : Tor C) (purine : A or G) A C 3'
3' C A (purine : A or G) (pyrimidine : T or C) T G 5'
They found that the Hind II enzyme always cuts directly in the center of this sequence.
Wherever this particular sequence of six base pairs occurs unmodified in a DNA molecule, Hind II will cleave both DNA strands or backbones between the 3rd and 4th base pairs of the sequence.
Moreover, Hind II will only cleave a DNA molecule at this particular site. For this reason, this specific base sequence is known as the "recognition sequence" for Hind II.
Hind II is just one example of the class of enzymes known as restriction nucleases.
In fact, more than 900 restriction enzymes, some sequences specific and some not, have been isolated from over 230 strains of bacteria since the initial discovery of Hind II.
These restriction enzymes generally have names that reflect their origin.
The first letter (in italics) of the name comes from the genus and the second two letters (in italics) come from the species of the prokaryotic cell from which they were isolated.
Next is the strain of the organism and last is the roman numeral indicating the order of discovery.
For example, EcoRI comes from Escherichia coli RY strain and was the first endonuclease isolated from bacteria, while Hind II comes from Haemophilus influenzae strain Rd. Numbers following the nuclease names indicate the order in which the enzymes were isolated from single strains of bacteria.
Nucleases are further described by addition of the prefix "endo" or "exo" to the name: The term "endonuclease" applies to sequence specific nucleases that break nucleic acid chains somewhere in the interior, rather than at the ends of the molecule.
Nuclease that function by removing nucleotides from the ends of the molecules are called "exonucleases".
Three main classes of restriction endonucleases – type-I, type-II and type-III have been described, each distinguished by a slightly different mode of action.
Out of these three types, type-I and type-III restriction enzymes are not used in recombinant DNA technology.
Type-II restriction enzymes are used in recombinant DNA technology, because they can be used In vitro to recognise and cut within the specific DNA sequence typically consisting of 4-8 nucleotide.
Type-I enzymes recognize specific sites within the DNA but do not cut at these sites.
Hence heterogeneous population of DNA fragments is produced, and therefore, type I enzymes do not take part in the technology. Type-III enzymes recognise a specific sequence of DNA molecule. Thus, products of type-III enzymes are homogeneous population of DNA fragments, so they cannot be used for genetic engineering experiments.
Table-I : Recognition Sequences of Several Restriction Endonucleases
(arrow indicates the site of cleavage)

The DNA segments cut by restriction enzymes are palindromic i.e., the nucleotide sequence of these DNA pieces read the same both, backwards and forward when orientation of reading is kept same, e.g., madam.
Blunt or flush ends are produced by many restriction enzymes which cleave both stands of DNA at exactly the same nucleotide position, in the centre of recognition site. For example, Sma I recognises 6 nucleotide palindromic sequence.
5'-C-C-C-G-G-G-3'
3'-G-G-G-C-C-C-5'
It cuts both DNA strands producing blunt ends.
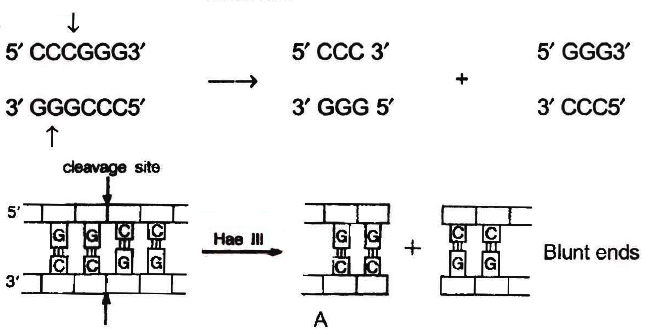
Action of restriction enzymes : The nucleotide sequences recognised and
cut by widely used restriction endonucleases A. Hae III
Sticky or cohesive ends are produced when restriction enzymes do not cut DNA at the same nucleotide position but cut the recognition sequence unequally. This produces short, single-stranded overhangs at each end. These are known as sticky ends. For example, Eco RI recognises 6 nucleotide palindromic sequence.
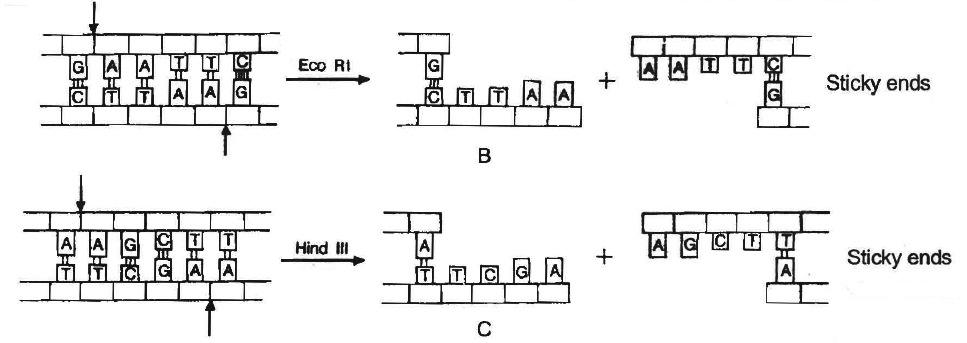
Action of restriction enzymes : The nucleotide sequences recognised and
cut by widely used restriction endonucleases B. Eco RI, C. Hind IIIC
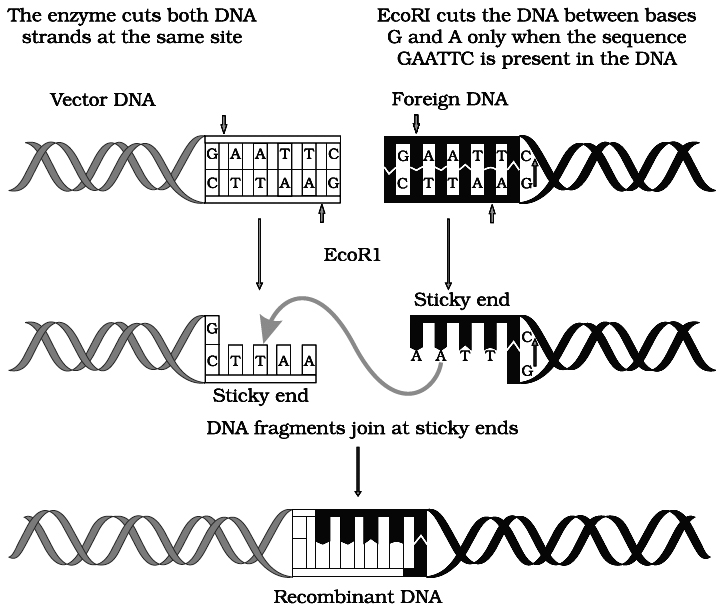
Action of restriction enzyme Eco RI and formation of recombinant DNA.
 This restriction endonuclease cuts both DNA strands unequally, producing 5' overhangs of 4 nucleotides.
This restriction endonuclease cuts both DNA strands unequally, producing 5' overhangs of 4 nucleotides.
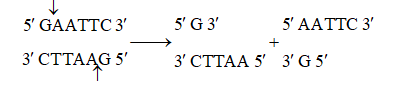
The stickiness helps enzyme ligase to make the DNA pieces join.
Other Enzymes used in Recombinant DNA Technology
In addition to restriction enzymes, there are several other enzymes that play an important role in DNA technology.
Three of the important ones are DNA ligase, alkaline phosphatase and DNA polymerase.
(a) DNA Ligase : This enzyme forms phosphodiester bonds between adjacent nucleotides and covalently links two individual fragments of double-stranded DNA. The action of the ligase enzyme requires a phosphate group at the 5' carbon of one nucleotide and a hydroxyl group at the 3' carbon of the adjacent nucleotide to form the phosphodiester bond between these two nucleotides. The enzyme used most often in the rDNA technology is T4 DNA ligase, which is encoded by phage T4.
(b) Alkaline Phosphatase (AP) : As mentioned above, ligation absolutely requires the presence of 5' phosphate group at the DNA site to be ligated. If this phosphate group is removed, this DNA cannot be ligated. The enzyme alkaline phosphatase is used to remove the phosphate group from the 5' end of a DNA molecule, leaving a free 5' hydroxyl group. This enzyme can be isolated from bacteria (BAP) or calf intestine (CAP). It is used to prevent unwanted self-ligation of vector DNA molecules in procedures of rDNA technology. However, ligation of the vector to the insert can occur as the insert still has its 5' phosphate.
(c) DNA Polymerase : DNA Pol I enzyme polymerizes the DNA synthesis on DNA template or complementary DNA (cDNA). It also catalyses a 5' 3' and 3' 5' exonucleolytic degradation of DNA. The other two enzymes are DNA polymerase II (DNA pol II) and DNA polymerase III (DNA pol III). These have almost similar catalytic activity. DNA pol III is about several times more active than the other two. Where there is preformed DNA template, it produces a parallel strand in the presence of ATP.
Separation and Isolation of DNA Fragments
After the cutting of DNA by restriction enzymes, fragments of DNA are formed.
These fragments can be separated by a technique called gel electrophoresis.
Electrophoresis is a technique of separation of charged molecules under the influence of an electrical field so that they migrate in the direction of electrode bearing the opposite charge, viz., positively charged molecules move towards cathode (-ve electrode) and negatively charged molecules travel towards anode (+ve electrode) through a medium/matrix.
This technique was developed by A. Tiselius in 1937.
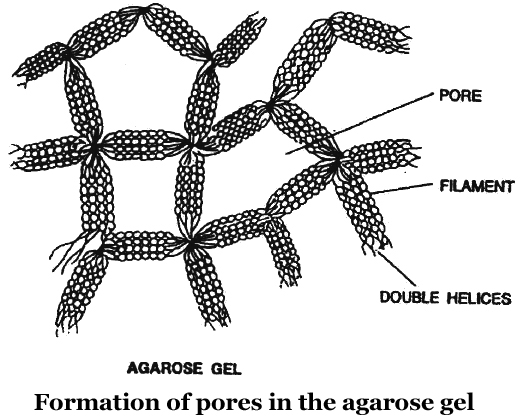
Now a days, the most commonly used matrix is agarose which is a polysaccharide extracted from sea weeds.
DNA fragments separate according to size through the pores of agarose gel.
Hence, the smaller the fragment size, the farther it moves.
Agarose dissolve in hot water, when this solution is cooled, double helices form and become arranged laterally and produce thick filaments.
These filaments become cross-linked to form the gel.
Pore size depends on agarose concentration.
The separated DNA fragments can be seen only after staining the DNA with a compound known as ethidium bromide followed by exposure to UV radiation as bright orange coloured bands. The separated bands of DNA are cut out from the agarose gel and extracted from the gel piece. This step is called as elution. Several techniques are used for eluting the DNA from the gel piece. These purified DNA fragments are used in the formation of recombinant DNA by linking them with cloning vectors.
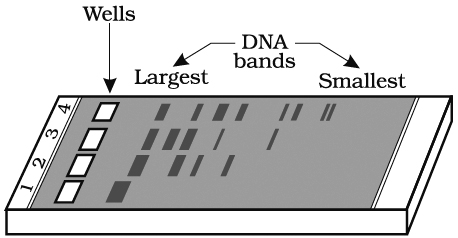
and digested set of DNA fragments (lane 2 to 4)
Cloning Vectors : Another important tool for genetic engineering is the vehicle for cloning, called vector. The vector carries a foreign DNA sequence into a given host cell. Bacterial plasmids and bacteriophages are considered the most useful. This is because :
(1) These are independent of the control of chromosomal DNA,
(2) Bacteriophage genomes occur in very large numbers in bacterial cells.
(3) The copies of plasmids per cell range from only a few to hundred or even more.
Certain essential features should be present in a DNA molecule to act as a cloning vector.
1. Origin of replication (ori): This is a DNA sequence which serves as a starting point for replication. When a DNA fragment gets associated with ori, foreign DNA into the vector would also replicate inside the host cell. Some vectors possess origin which favours formation of high copy numbers and hence preferred.
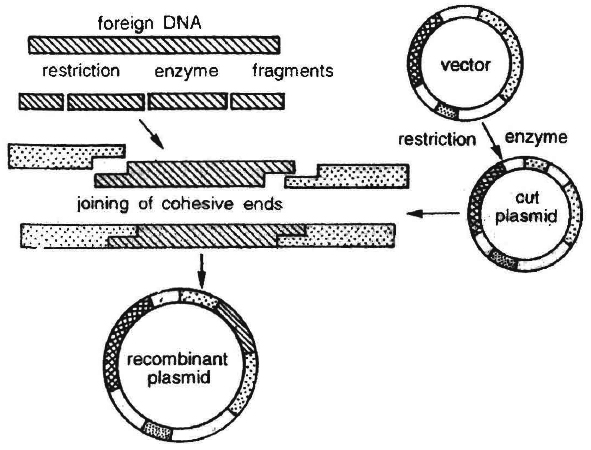
2. Selectable marker: Vector should also include a selectable marker. This is a gene which would permit the selection of host cells containing vector from amongst those which do not possess vector. Common selectable markers include genes encoding antibiotic resistance such as ampicillin resistance or enzymes such as -galactosidase (product of lac Z gene of lac operon). These genes can be identified by a colour reaction.
3. Recognition sites : Vector should possess an unique restriction site that would allow particular enzyme to cut the vector only once. This site would be recognised by the commonly used restriction enzymes. If there are more than one recognition site in the vector, several fragments would be produced. Generally the vectors used possess unique recognition sites for several restriction enzymes in a small region of DNA. This is known as polylinker or multiple cloning site (MCS). Such a cloning site offers choice of restriction enzyme.
Unique restriction endonuclease recognition site enables insertion of foreign DNA into the vector for production of recombinant DNA. The foreign DNA is inserted and made to join (ligate) at a specific restriction site generally in antibiotic resistance gene.
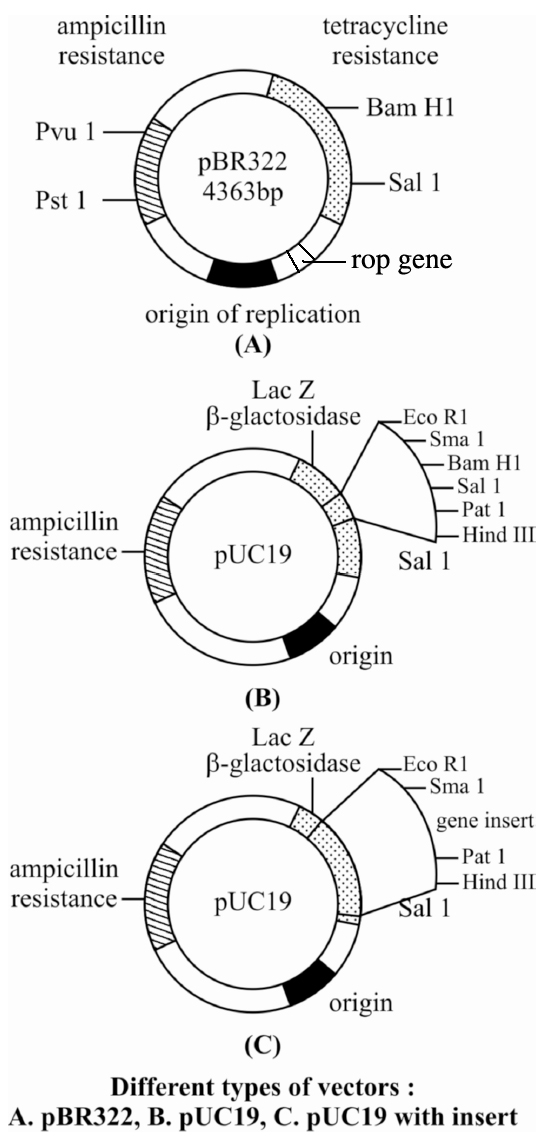
pBR322 has genes for resistance against two antibiotics tetracycline and ampicillin, an origin of replication and a variety of restriction sites for cloning of restriction fragments obtained through cleavage with a specific enzyme. Foreign DNA is inserted at a site located in one of the two genes for resistance against antibiotics, so that it will inactivate one of the two resistance genes. The insert bearing plasmid can be selected by their ability to grow in a medium containing only one of the two antibiotics and their failure to grow in a medium containing both antibiotics. The plasmids carrying no insert on the other hand, will be able to grow in a media containing one or both the antibiotics. In this way, the presence of resistance genes against ampicillin and tetracycline allow selection of Escherichia coli colonies transformed with plasmids carrying the desired foreign cloned DNA fragment.
4. Size of the vector : Cloning vector should be small in size. Large molecules have a tendency to breakdown during purification. These are also difficult to manipulate.
DIFFERENT TYPES OF VECTOR
Several types of vectors satisfying the above characters have been developed. The following are some of the commonly used vectors.
1. Plasmids:
These are extra-chromosomal, non-essential self-replicating, usually circular and double stranded DNA molecules occurring in some bacteria and also a few yeasts.
Some of the characters carried by plasmid may not be required for normal bacterial metabolism but may be of great advantage e.g. antibiotic resistance.
pBR322 is one of the standard cloning vectors widely used in gene cloning experiments.
This vector has been restructured by inserting genes for antibiotic resistance.
It is named after Boliver and Rodriguez who prepared this vector, pUC (named after University of California) is another such reconstructed plasmid vector.
The vectors mentioned above are able to replicate only in E. coli.
Therefore, many vectors constructed for eukaryotic cells are also functional in E. coli.
These vectors are called shuttle vectors.
The vectors contain two types of origin of replication and selectable marker genes-one for the eukaryotic cell and another for E. coli.
The common example of this type is yeast episomal plasmid (YEp).
In plants, Ti plasmid of bacterium Agrobacterium tumfaciens has been modified to function as vector.
2. Vectors based on bacteriophages :
Bacteriophages are viruses which infect bacterial cells produce new phages inside the host bacterium, and are released from the host cell to again infect other bacterial cells. M 13 and lambda () phages are in common use.
3. Cosmids:
These combine some features of plasmid and 'cos' (cohesive end sites) of phage lambda (cosmid = cos + plasmid).
4. YAC vectors:
Yeast artificial chromosome contain telomeric sequence, the centromere and autonomously replicating sequence from yeast chromosomes. These also have suitable restriction enzyme sites and genes useful as selectable markers.
5. BAC vectors:
Bacterial artificial chromosome is based on F plasmid (fertility) of E. coli. It contains genes for replication and maintenance of F-factor, selectable marker and cloning sites.
Colour Reaction : Due to inactivation of antibiotics, selection of recombinants becomes burdensome process because it requires simultaneous plating on two plates having different antibiotics. Thus, alternative selectable marker is developed to differentiate recombinants and non-recombinants on the basis of their ability to produce colour in the presence of a chromogenic substance. Now a recombinant DNA is inserted in the coding sequence of an enzyme -galactosidase. This causes inactivation of the enzyme which is called insertional inactivation. If the plasmid in the bacterium does not have an insert, the presence of a chromogenic substrate gives blue coloured colonies. Presence of insert results into insertional inactivation of the -galactosidase and, therefore, the colonies do not produce any colour, these colonies are marked as recombinant colonies.
6. Vectors for Cloning Genes in Plants and Animals:
We know the procedure of transferring genes into plants and animals from bacteria and viruses.
It is also known how to transfer genes to transform eukaryotic cells and force them to do what the bacteria or viruses require.
For example, Agrobacterium tumifaciens, a pathogen (disease causing agent) of several dicot plants is able to transfer a piece of DNA known as 'T-DNA' to convert normal plant cells into tumour and direct these tumour cells to secrete the chemicals required by the pathogen.
Similarly, retroviruses (cause leukosis or sarcoma types of cancer) in animals including humans are able to change normal cells into cancerous cells.
The tumour inducing (Ti) plasmid of Agrobacterium tumifaciens has been modified into cloning vector which is not pathogenic to the plants, however, it is still able to use the procedure to deliver genes of our interest into various plants.
Similarly retroviruses are used to carry desirable genes into animal cells.
Thus once a gene or DNA fragment is joined to a suitable vector it is transferred into a bacterial plant or animal host where it undergoes multiplication.
HOST CELL
Competent host cell is required for transformation with recombinant DNA.
After formation of recombinant DNA, propagation of it must occur inside a living system or a host.
Different type of available host cells are like E. coli, yeast, animal and plant cells.
The type of host cell to be used depends on the aim of cloning experiment.
Eukaryotic cells will be the preferred host for expression of some eukaryotic proteins.
Yeast cells are preferred because these are simplest eukaryotic organisms and like bacteria are single celled, genetically well characterized, easy to grow and manipulate.
Plant and animal cells can be used for protein expression either in tissue culture or as cells in the whole organism to create genetically modified (GM) crops and animals.
As DNA is hydrophilic molecule, it can not pass through cell membrane.
Therefore, the bacterial cells should be capable of uptaking DNA.
This is accomplished by treating them with specific concentration of a divalent cation, e.g., Ca2+ thus making them competent which causes efficient entry of DNA into bacterium through pores in its cell wall.
Recombinant DNA can be forced into such cells by incubating the cells with recombinant DNA on ice, followed by placing them briefly at 42°C (heat shock) and then putting them back on ice. As a result, bacteria gets enabled to pick up recombinant DNA.
There are other methods to introduce foreign DNA into host cells. These are briefly described below.
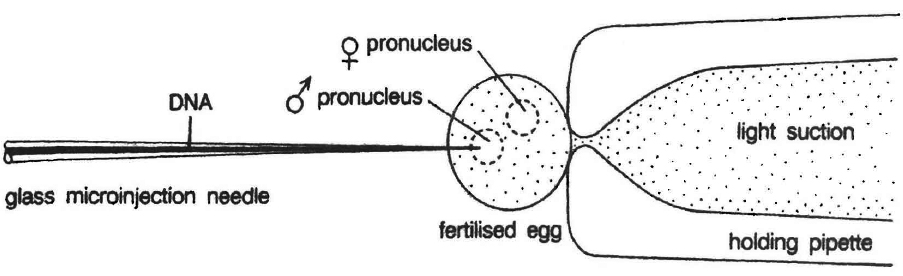
Microinjection
In this method recombinant DNA is directly injected into the nucleus of animal cell by using micro needles or micro pipettes. It is used in oocytes, eggs and embryo. Jeffey S. Chamberlain et. al. (1993) of Human Genome Centre, Michigan University, USA have cured mice that inherited a neuromuscular disease which is like muscular dystrophy of humans.
Direct DNA Injection
Direct injection of DNA into skeletal muscle led to the possibility of using gene as vaccines. Due to low level of expression, therapeutic benefits for the treatment of genetic disorder could not be derived. This method gave birth to the concept of DNA vaccine or genetic immunization.
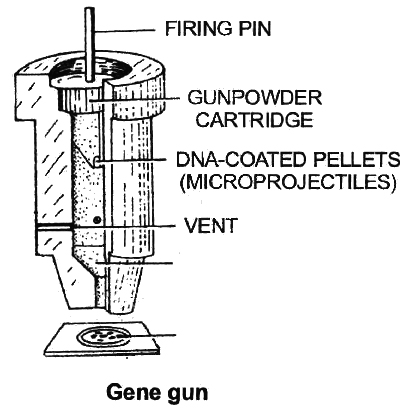
Gene Gun or Biolistics
New technologies like gene gun are also available for vectorless direct gene transfer. DNA coated onto microscopic pellets is literally shot into target cells. Although, it is developed for plants but is also used for animal cells for promoting tissue repair or reducing healing time. This method made great impact in the field of vaccine development.
processes of recombinant DNA technologies
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
PROCESS OF RECOMBINANT DNA TECHNOLOGY
Recombinant technology is a complicated process. Several steps lead to the desired goal. The major steps are:
(1) Isolation of DNA,
(2) Digestion of DNA by restriction endonuclease enzyme,
(3) Gene amplification,
(4) Introduction of recombinant DNA into host cells,
(5) Identification of recombinants,
(6) Gene product manufacture, and
(7) Processing.
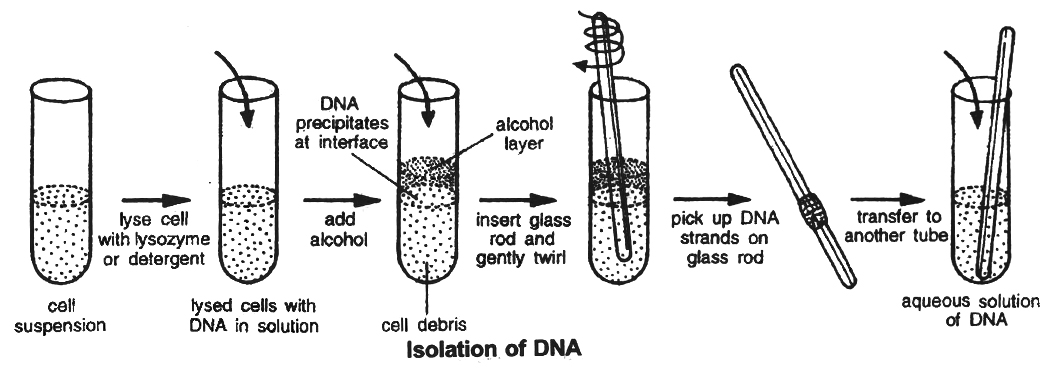
(1) Isolation of DNA: Isolation of the Genetic Material (DNA)
Nucleic acid (DNA or RNA) is the genetic material of all organisms. It is DNA in majority of organisms.
For cutting the DNA with restriction enzymes it needs to be pure and free from other macromolecules.
Because the DNA is covered by the membranes, it has to break the cell open to release DNA and other macromolecules like RNA, proteins, polysaccharides and lipids.
It is obtained by treating the bacterial cells/plant or animal tissue with enzymes such as lysozyme (bacteria), cellulase (plant cells), chitinase (fungus).
As we know that genes are present on long molecules of DNA intertwined with proteins like histones, the RNA can be removed by treating with ribonuclease while proteins can be removed by treating with protease.
Other molecules are removed by proper treatments. The purified DNA finally precipitates out after the addition of chilled ethanol.
This is seen as collection of fine threads in suspension.
(2) DNA digestion by restriction enzymes
The vector and the target DNA fragment can be separately digested with the same restriction enzyme.
The digested vector and the target DNA fragment are then incubated together in the presence of DNA ligase enzyme.
Incubation results in bonding two types of DNA by phosphodiester bonds between them.
Thus, deoxyribose-phosphate backbones of vector molecule and the target DNA fragment are covalently linked, forming a recombinant DNA molecule.
Another possibility in this experiment is the rejoining of the sticky ends of the vector molecule itself, forming a circular vector DNA molecule that is without foreign DNA molecule.
This possibility is eliminated by treating digested, vector with alkaline phosphatase or by using different restriction enzymes.
(3) Gene amplification
This is the process of selective multiplication of a specific region of DNA molecule.
The process has also been used to produce DNA fragments for cloning.
Amplification is achieved by a special method known as polymerase chain reaction (PCR) developed by Kary Mullis in 1985 for which he shared 1993 Nobel Prize.
The principle underlying the technique is to heat double stranded DNA molecule to a high temperature so that the two DNA strands separate into single stranded DNA molecules.
If these single-stranded molecules are copied by a DNA polymerase, it would lead to duplication of the original DNA molecule and if these events are repeated many times, multiple copies of the original DNA sequence can be generated.
The basic requirements of a PCR reaction are the following:
(i) DNA Template: Any source that contains one or more target DNA molecules to be amplified can be taken as template.
(ii) Primers : Primers, which are oligo-nucleotides, usually 10-18 nucleotides long, that hybridize to the target DNA region, one to each strand of the double helix. Two primers are required and these primers are oriented with their ends facing each other, allowing synthesis of the DNA towards one another.
(iii) Enzyme: DNA polymerase which is stable at high temperatures (>90º) is required to carry out the synthesis of new DNA. The polymerase which is generally used in PCR reactions is known as Taq polymerase (isolated from a bacterium Thermus aquaticus). Other thermostable polymerases can also be used.
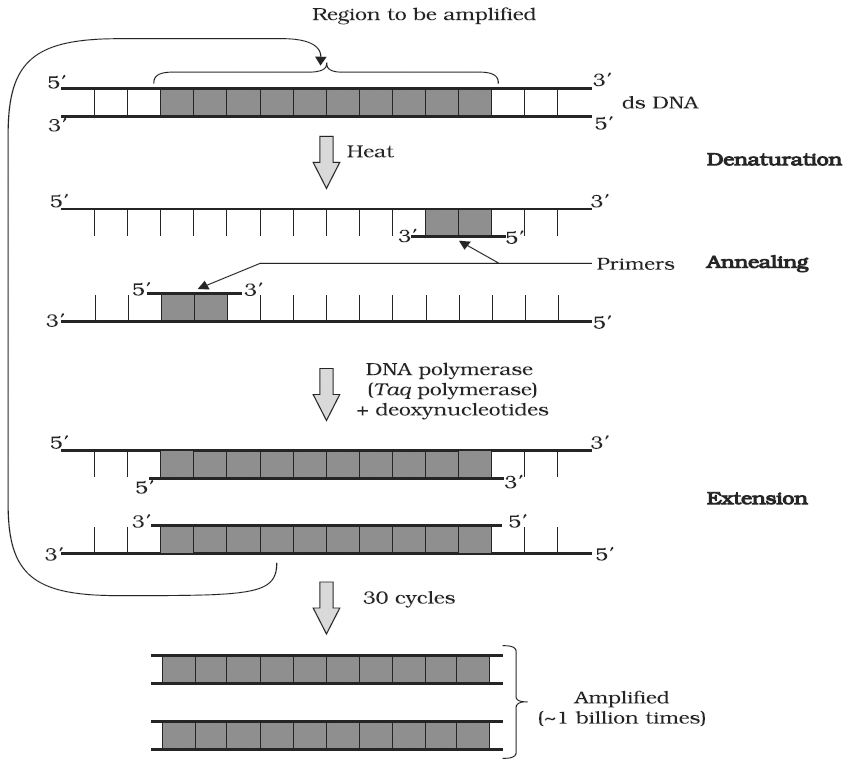
A schematic representation of the three steps performed during PCR. Note that the two primers used are complementary to the 3' end sequences of DNA segment to be amplified
Working Mechanism of PCR :
A single PCR amplification cycle involves three basic steps denaturation, annealing and extension (Polymerisation).
(a) Denaturation: In the denaturation step, the target DNA is heated to a high temperature (usually 94°C), resulting in the separation of the two strands. Each single strand of the target DNA then acts as a template for DNA synthesis.
(b) Annealing: In this step, the two oligo-nucleotide primers anneal (hybridize) to each of the single stranded template DNA since the sequence of the primers is complementary to the 3' ends of the template DNA. This step is carried out at a lower temperature depending on the length and sequence of the primers.
(c) Primer Extension (Polymerisation) : The final step is extension, wherein Taq DNA polymerase (of a thermophilic bacterium Thermus acquaticus) synthesizes the DNA region between the primers, using dNTPs (deoxynucleoside triphosphates) and Mg2+. It means the primers are extended towards each other so that the DNA segment lying between the two primers is copied. The optimum temperature for this polymerization step is 72°C.
To begin the second cycle, the DNA is again heated to convert all the newly synthesized DNA into single strands, each of which can now serve as a template for synthesis of more new DNA. Thus the extension product of one cycle can serve as a template for subsequent cycles and each cycle essentially doubles the amount of DNA from the previous cycle. As a result, from a single template molecule, it is possible to generate 2n molecules after n number of cycles.
Concept Builder
PCR permits early diagnosis of malignant diseases such as leukemia and lymphomas, which is currently the highest developed in cancer research and is already being used routinely.
PCR assays can be performed directly on genomic DNA samples to detect translocation specific malignant cells at a sensitivity which is at least 10,000 fold higher than other methods.
PCR also permits identification of non-cultivatable or slow-growing microorganisms such as mycobacteria, anaerobic bacteria, or viruses from tissue culture assays and animal models.
The basis of PCR diagnostic applications in microbiology is the detection of infectious agents and the discrimination of non-pathogenic from pathogenic strains by virtue of specific genes.
Viral DNA can likewise be detected by PCR.
The primers used need to be specific to the targeted sequences in the DNA of a virus, and the PCR can be used for diagnostic analyses or DNA sequencing of the viral genome.
The high sensitivity of PCR permits virus detection soon after infection and even before the onset of disease.
Such early detection may give physicians a significant lead in treatment.
The amount of virus ("viral load") in a patient can also be quantified by PCR-based DNA quantitation techniques.
Application of PCR
Some of the areas of application of PCR are briefly mentioned here.
(i) Diagnosis of Pathogens : Pathologists use techniques based on detecting specific enzymes or antibodies against disease-related proteins. But these techniques cannot be used for detecting infectious agents that are difficult to culture or that persist at very low levels in infected cells. To overcome these problems, PCR-based assays have been developed that detect the presence of gene sequences of the infectious agents.
(ii) Diagnosis of specific Mutation : PCR can be used to detect the presence of a specific mutation that is responsible for causing a particular genetic disease before the actual onset of the disease. By using PCR, phenylketonuria, muscular dystrophy, sickle cell anaemia, AIDS, hepatitis, chlamydia and tuberculosis can be diagnosed.
(iii) DNA Finger printing: PCR is of immense value in generating abundant amount of DNA for analysis in the DNA fingerprinting technique used in forensic science to link a suspect's DNA to the DNA recovered at a crime scene.
(iv) Detection of Specific Microorganisms: PCR is also used for detecting specific microorganisms from the environment samples of soil, sediments and water.
(v) In Prenatal Diagnosis: It is useful to detect genetic disease in foetus before birth. If the disease is not curable, abortion is recommended.
(vi) Diagnosis of Plant Pathogens : Many diseases of plants can be detected by using PCR. For examples, viroids (associated with apple, grape, citrus, pear, etc.), viruses (like TMV, bean yellow mosaic virus etc.), bacteria, mycoplasmas, etc.
(vii) In Palaeontology: PCR is used to clone the DNA fragments from the mummified remains of humans and extinct animals like wooly mammoth and dinosaurs.
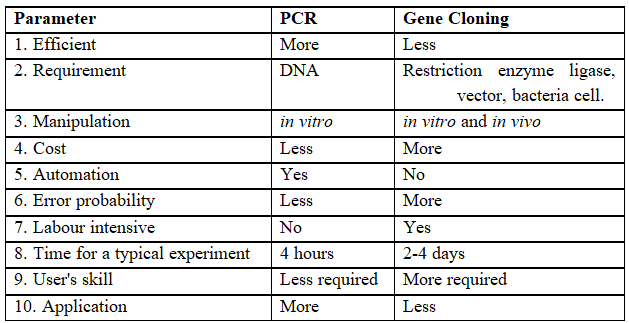
Table-II : Comparison between PCR and Gene Cloning

Concept Builder
Definition of Ligase chain reaction (LCR)
A method of DNA amplification similar to PCR.
LCR differs from PCR because it amplifies the probs molecule rather than producing amplicon through polymerization of nucleotides.
Two probes are used per each DNA strand and are ligated together to 'form' a single probe.
LCR uses both a DNA polymerase enzyme and a DNA ligase enzyme to drive the reaction.
Like PCR, LCR requires a thermal cycler to drive the reaction and each cycle results in a doubling of the target nucleic acid molecule. LCR can have greater specificity than PCR.
Polymerase Chain Reaction (PCR) : Technique for obtaining multiple copies of DNA
(4) Introduction of recombinant DNA into host cells:
Once the recombinant DNA molecule has been multiplied, it needs to be inserted into host cells.
Many methods for introduction are available.
Selection of a method depends upon type of vector and the host cell along with other things.
Some common methods are outlined below-
(a) Transformation: This is a method where cells take up DNA from their surroundings. Since, many cells such as those of E.coli, yeast, mammalian cells, etc. do not naturally absorb DNA, hence they need to be made competent. Mandel and Higa (1970) observed that E. coli cells can be made competent to take up external DNA by suspending them in cold calcium chloride.
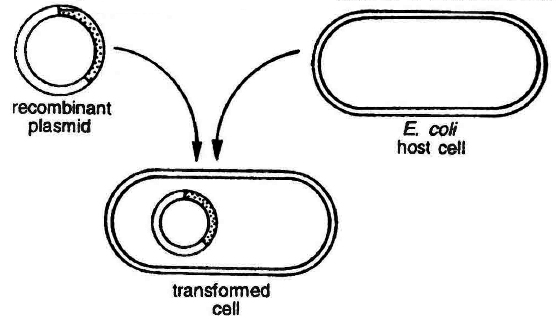
Transformation in bacterial cell
(b) Transfection: In this method DNA is mixed with charged substances like calcium phosphate, cationic liposomes etc. These are spread on the recipient host cells. Calcium ions carry foreign DNA and release it inside the cell since calcium gets precipitated in the form of calcium phosphate, thus transferring the DNA by endocytosis.
(c) Microinjection and macroinjection: Specially designed micromanipulator is used to inject DNA into cytoplasm or the nucleus of a recipient cell or protoplast. The method is used for direct introduction of DNA into plant or animal cells without using special eukaryotic vectors.
(d) Microprojection (biolistics or particle gun) : Tungsten or gold particles (microparticles) coated with DNA are accelerated to a very high initial velocity. These microprojectiles are carried by other (nylon) microprojectiles or the bullet, causing them to penetrate the cell walls of intact target cells or tissues.
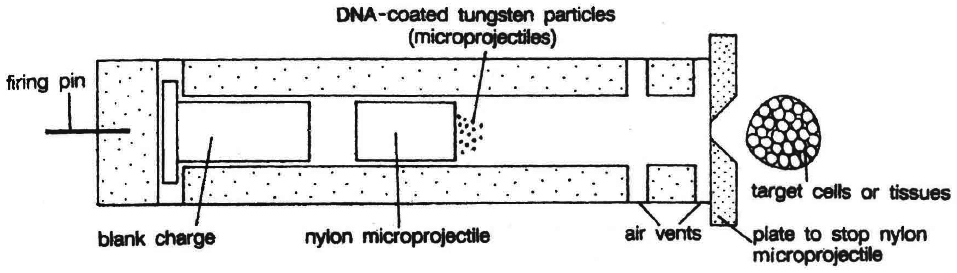
(e) Electroporation: Short electrical impulses of high field strength are given. These increase the permeability of protoplast membrane by creating transient microscopic pores, thus making the entry of DNA molecules into the cells much easier.
(f) Ti plasmid based gene transfer: A more common method of introducing foreign DNA into plant cells is to use the bacterium Agrobacterium tumefaciens and its Ti plasmid.
This gram negative soil bacterium is a plant pathogen and produces crown gall disease in many dicotyledons including grapes, stone fruits, roses, tomato, sunflower, cotton, soyabean, etc.
Most strains of this bacterium carry tumour inducing (Ti) plasmid.
In nature, Agrobacterium attaches to the leaves of the plants and Ti plasmid is transferred into plant cells.
The plasmid becomes incorporated into plant chromosomal DNA.
Therefore, Ti plasmid has been used as a vehicle for introduction of recombinant DNA into plant cells.
Ti plasmids cause tumours in plants.
Strains of the bacterium have been developed which do not have tumour inducing genes.
However, T region of plasmid plays an important role in gene transfer.
This specific segment of bacterial plasmid DNA is called T-DNA (transferred DNA).
T-DNA has a cloning site into which foreign DNA (DNA insert) is inserted.
This recombinant plasmid is now introduced into the bacterium Agrobacterium tumefaciens.
It is then used to infect cultured cells.
The T region of the plasmid with foreign DNA (or DNA insert) is transferred to plant cells.
It gets integrated with chromosomal DNA of the cell.
Cultured cells are induced to grow into plantlets.
These are planted into the soil where the mature plants are formed.
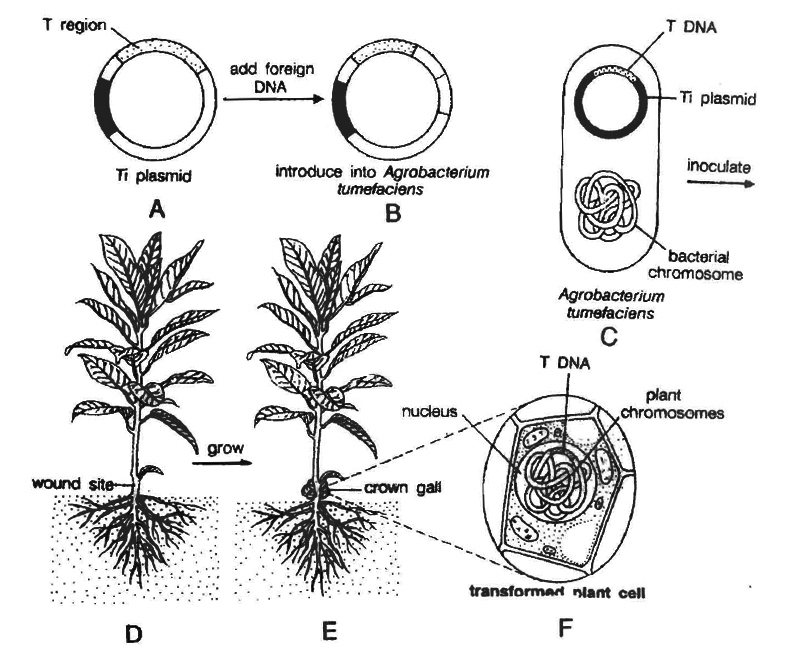
(5) Identification of recombinant :
After the insertion of recombinant DNA into the host cell, these need to be identified from those which do not possess it.
The methods used to do so consider expression or non-expression of certain characters especially antibiotic resistance-gene (e.g., ampicillin resistance gene) on plasmid vector.
Selectable marker usually provides resistance against a substrate which when added to the culture medium, inhibits the growth of normal cells or tissues in culture, so that only transformed tissues will grow.
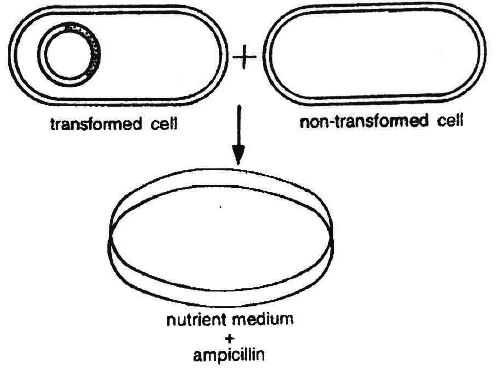
Thus the simplest method for identification is to grow transformed host cells (with ampicillin resistance gene) on medium containing ampicillin.
This would enable the cells containing this transformed plasmid to grow and form colonies.
There are other methods for detection of recombinants based on the fact that the cloned DNA fragment disturbs the coding sequence of gene.
This is known as insertional inactivation.
Let us consider a plasmid containing genes resistant for two different antibiotics, i.e., ampicillin and tetracycline.
If the target DNA fragment is inserted in a site located in ampicillin resistance gene, this gene will then be inactivated.
Thus, host cells with such a recombinant plasmid will be sensitive to ampicillin but resistant to tetracycline.
These host cells will die when grown on ampicillin containing medium but would grow on medium containing tetracycline.
Self ligated or religated (non-recombinant) vectors would grow on medium containing both ampicillin and tetracycline being resistant to them.
Another, but similar method involves insertional inactivation of lac Z gene.
It is known as blue-white selection, being colour based.
(6) Gene product manufacture:
When recombinant DNA is transferred into a bacterial, plant or animal cell, the foreign DNA is multiplied.
Most of the recombinant technologies are aimed to produce a desirable protein.
So there is a need for expression recombinant DNA.
After the cloning of the gene of interest one has to maintain the optimum conditions to induce the expression of the target protein and consider producing it on a large scale.
If any protein encoding gene is expressed in a heterologous host it is known as a "recombinant protein".
The cells having cloned genes of interest can be grown on a small scale in the laboratory.
The cultures may be used for extracting and purifying the desired protein.
The cells can also be multiplied in a continuous system where the used medium is passed out from one side and fresh medium is added from the other side to maintain the cells in their physiologically most active lag exponential phase (Lag phase -no significant increase of the cells, exponential phase -rapid multiplication of the cells).
This type of culturing method produces a larger biomass to get higher yields of desired protein.
Small volume cultures cannot give large quantities of the products.
To produce large quantities of these products, development of "bioreactors" was required where large volumes (100-1000 litres) of culture can be processed, Hence, bioreactors are like vessels in which raw materials are biologically converted into specific products, individual enzymes using microbial, plant, animal or human cells.
A bioreactor provides the optimal conditions for obtaining the desired product by providing optimum growth conditions such as substrate, temperature, pH, vitamins, oxygen and salts.
One of the most commonly used bioreactor is of stirring type.
The presence of stirrer makes mixing possible and also makes oxygen available through the reactor.
A bioreactor also has an agitatory system, an oxygen delivery system, a foam control system, a temperature control system, pH control system and sampling ports so that small volumes of the culture can be withdrawn periodically.
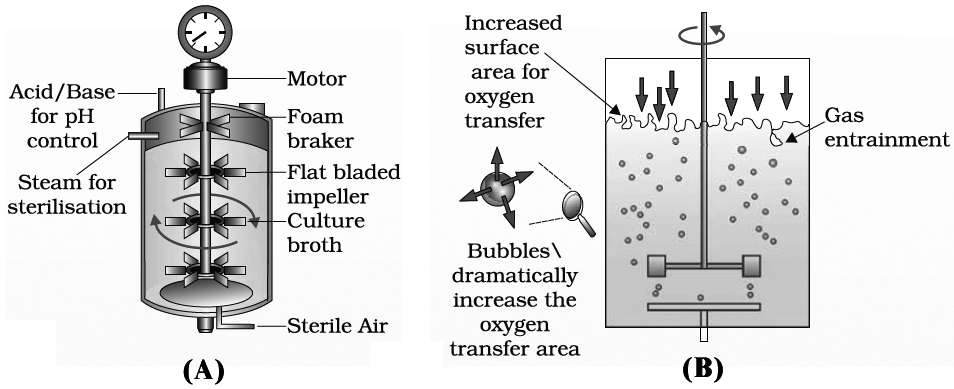
which sterile (free from any germs) air bubbles are sparged.
(7) Downstream Processing :
Once the product is ready, it has to be processed for commercial use.
This requires purification and strict quality control to maintain the efficacy.
The products based on biotechnology must ensure that they satisfy the consumer needs and are not harmful.
Therefore, a thorough checking of products at each level of manufacture is done.
The manufacturing process and the quality control methods vary with each product.
Biotechnological applications in agriculture
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
BIOTECHNOLOGICAL APPLICATION IN AGRICULTURE
Food production can be enhanced by
(1) Agro-chemical based Agriculture
(2) Organic agriculture
(3) Genetically engineered crop-based agriculture
Green revolution resulted in increasing the food supply almost three times.
Green revolution is the great increase in the production of food grains (especially wheat and rice) that resulted in large part from the introduction of new, high yielding varieties begining in the mid 20th century.
Its early dramatic success was Maxico and the Indian subcontinent.
The new varieties required large amount of chemical fertilizers and pesticides to produce high yields, raising concern about cost and potentially harmful environmental effects.
This demands an alternate pathway that can result in maximum yield from the fields but the chemicals and fertiliser use is minimum i.e., harmful effects on the environment are reduced.
Genetically modified organisms or GMO can be the plants, bacteria, fungi and animals whose genes have been alteres by genetic manipulation.
(A) Genetically Modified Crops :
A transgenic crop is a crop that contains and expresses a transgene.
A popular term for transgenic crops is genetically modified crops or GM crops.
The techniques used for the production of transgenic crops offer the following two unique advantages: (i) any gene (from any organism or a gene synthesised chemically) can be used for transfer, and (ii) the change in genotype can be precisely controlled since only the transgene is added into the crop genome.
In contrast, breeding activities
(i) Can use only those genes that are present in such species that can be hybridised within them. In addition,
(ii) Changes occur in all those traits for which the parents used in hybridisation differ from each other.
When a transgene is introduced into the genome of an organism, it can achieve one of the following :
(i) Produces a protein that is the product in which we are interested.
(ii) Produces a protein that on its own produces the desired phenotype.
(iii) Modifies an existing biosynthetic pathway so that a new end-product is obtained.
(iv) Prevents the expression of an existing native gene.
Hirudin is a protein that prevents blood clotting. The gene enconding hirudin was chemically synthesised. This gene was then transferred into Brassica napus, where hirudin accumulates in seeds. The hirudin is purified and used as medicine. In this case, the transgene product itself is the product of interest.
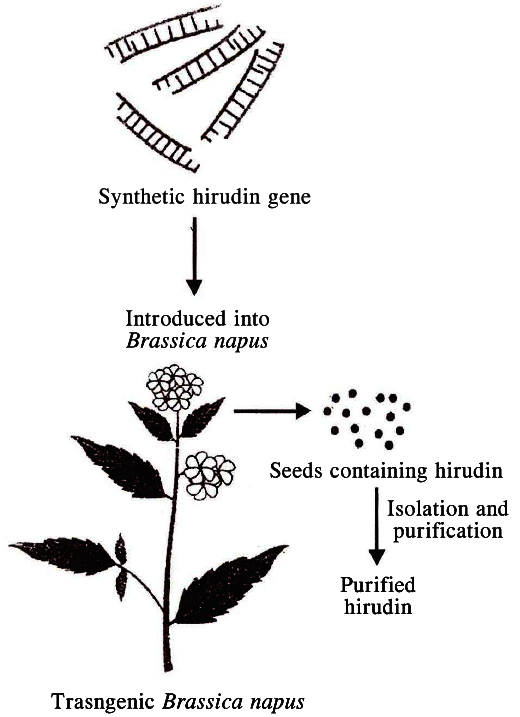
A simplified representation of the production of
hirudin from transgenic Brassica napus seeds
The tomato variety 'Flavr Savr' presents an example where expression of a native tomato gene has been blocked.
Expression of a native gene can be stopped by many different methods.
Fruit softening is promoted by the enzyme polygalacturonase which degrades pectin.
Production of polygalacturonase was blocked in, the transgenic tomato variety 'Flavr Savr'.
Therefore, fruits of this tomato variety remain fresh and retain their flavour much longer than do the fruits of normal tomato varieties. In addition, the fruits have a superior taste and increased total soluble solids these are unexpected bonus.
(B) Genetically Modified Food:
The food prepared from the produce of genetically modified (= transgenic) crops is called genetically modified food or, in short, GM food. GM food differs from the food prepared from the produce of conventionally developed varieties mainly in the following aspects.
Firstly, it contains the protein produced by the trans-gene in question, e.g., Cry protein in the case of insect resistant varieties.
Secondly, it contains the enzyme produced by the antibiotic resistance gene that was used during gene transfer by genetic engineering.
Finally, it contains the antibiotic resistance gene itself.
Concept Builder
Biofortification is a method of breeding crops to increase their nutritional value.
This can be done either through conventional selective breeding, or through genetic engineering.
Biofortification differs from ordinary fortification because it focuses on making plant foods more nutritious as the plants are growing, rather than having nutrients added to the foods when they are being processed.
This is an improvement on ordinary fortification when it comes to providing nutrients for the rural poor, who rarely have access to commercially fortified foods.
As such, biafortifications seen as an upcoming strategy for dealing with deficiencies of micronutrients in the developing world.
It has been argued that the above features of GM foods could lead to the following problems when they are consumed.
Firstly, the transgene product may cause toxicity and/or produce allergies.
Secondly, the enzyme produced by the antibiotic resistance gene could cause allergies, since it is a foreign protein.
Finally, the bacteria present in the alimentary canal of the human could takes up the antibiotic resistance gene that is present in the GM food.
These bacteria would then become resistant to the concerned antibiotic.
As a result, these bacteria could become difficult to manage.
The scientists involved in the production of transgenic crops are addressing to these concerns.
Efforts are being made to use other genes in place of antibiotic resistance genes.
The toxic and allergenic actions of the trans-gene product can be adequately examined by detailed assays using suitable animal models.
GM PRODUCTS: BENEFITS AND CONTROVERSIES
Benefits
(1) Crops
(i) Enhanced taste and quality
(ii) Reduced maturation time
(iii) Increased nutrients, yields, and stress tolerance
(iv) Improved resistance to disease, pests, and herbicides
(v) New products and growing techniques
(2) Animals
(i) Increased resistance, productivity, hardiness, and feed efficiency
(ii) Better yields of meat, eggs, and milk.
(iii) Improved animal health and diagnostic methods
(3) Environment
(i) "Friendly" bioherbicides and bioinsecticides
(ii) Conservation of soil, water, and energy
(iii) Bioprocessing for forestry prroducts
(iv) Better natural waste management
(v) More efficient processing
(4) Society
Increased food security for growing populations
Controversies
Safety: Potential human health impact: allergens, transfer of antibiotic resistance markers, unknown effects Potential environmental impact: unintended transfer of transgenes through cross-pollination, unknown effects on other organisms (e.g., soil microbes), and loss of flora and fauna biodiversity.
Bt COTTON
DNA technology makes it possible to locate the genes that produces Bt proteins lethal to insects and transfer the gene into crop plants.
First scientists identify a strain of Bt that kills the targeted insect.
Then they isolate the gene that produces the lethal protein.
That gene is removed from the Bt bacterium and a gene conferring resistance to a chemical (usually antibiotic or herbicide) is attached that proves useful in later steps.
The Bt gene with the resistance gene-attached is inserted into plant cells.
These modified or genetically transformed cells are then grown into complete plant by tissue culture.
The modified plant produces the same lethal protein as produced by the Bt bacteria because plants now have the same gene.
Concept Builder
B. thuringiensis was first discovered in 1902 by Japanese biologist Shigetane Ishwatari.
In 1911, B. thuringiensis was rediscovered in Germany by Ernst Berliner, who isolated it as the cause of a disease called Schlaffsucht in flour moth caterpillars.
In 1976, Zakharyan reported the presence of a plasmid in a strain of B. thuringiensis and suggested the plasmid's involvement in endospore and crystal formation.
B. thuringiensis is closely related to B.cereus, a soil bacterium, and B.anthracis, the cause of anthrax: the three organisms differ mainly in their plasmids.
Like other members of the genus, all three are aerobes capable of producing endospores. Upon sporulation, B. thuringiensis forms crystals of proteinaceous insecticidal -endotoxin (called crystal proteins of Cry proteins), which are encoded by cry genes.
In most strains of B. thuringiensis the cry genes are located on the plasmid.
Cry toxins have specific activities against insect species of the orders Lepidoptera (moths and butterflies), diptera (flies and mosquitoes), coleoptera (beetles), hymenoptera (wasps, bees, ants and sawflies) and nematodes.
Thus, B. thurengiensis serves as an important reservoir of Cry toxins for production of biological insecticides and insect-resistant genetically modified crops.
When insects ingest toxin crystals, the alkaline pH of their digestive tract activates the toxin.
Cry inserts into the insect gut cell membrane, forming a pore. The pore results cell lysis and eventual death of the insect.
B. thuringiensis forms protein crystals during a particular phase of their growth.
These crystals contain a toxic insecticidal protein.
Why does this toxin not kill the Bacillus? Actually, the Bt toxin protein exists as inactive protoxin but once an insect ingests the inactive toxin, it is converted into an active form of toxin due to the alkaline pH of the gut which solubilises the crystals.
The activated toxin binds to the surface of midgut epithelial cells and creates pores that cause cell swelling and lysis and eventually cause death of the insect.
Bt is not harmful to humans, other mammals, birds, fish or beneficial insects.
Specific Bt toxin genes were isolated from Bacillus thuringiensis and incorporated into the several crop plants such as cotton.
The choice of genes depends upon the crop and the targeted pest, as most Bt toxins are insect-group specific.
The toxin is coded by a gene named cry. There are a number of them, for example, the proteins encoded by the genec cry I Ac and cry II Ab control the cotton bollworm, that of cry I Ab controls corn borer.
Although Bt genes have been introduced into tobacco, tomatoes, cotton, and other broadleaf Plants, gene transfer technology for corn is a recent achievement.

The development of corn plants expressing Bt proteins requires substantial changes in the Bt genes, including the creation of synthetic versions of the genes, rather than the microbial Bt gene itself.
Concept Builder
There are several advantages in expressing Bt toxins in transgenic Bt crops:
The level of toxin expression can be very high thus delivering sufficient dosage to the pest:
The toxin expression is contained within the plant system and hence only those insects that feed on the crop perish.
The toxin expression can be modulated by using tissue-specific promoters, and replaces the use synthetic pesticides in the environment: The latter observation has been well documented Worldwide.
PEST RESISTANT PLANTS
Root-knot nematodes are the most economically important group of plant-parasitic nematodes worldwide. They attack nearly every food and fiber crop grown, about 2,000 plant species in all.
The nematode invades plant roots, and by feeding on the roots' cells, they cause the roots to grow large form galls, or knots, damaging the crop and reducing its yields .
The most cost-effective and sustainable management tactic for preventing root-knot nematode damage and reducing growers' losses is to develop resistant plants that prevent the nematode from feeding on the roots.
Because root-knot nematode resistance doesn't come naturally in most crops, bioengineering is required.
Four common root-knot nematode species (mainly Meloidegyne incognitia) account for 95 percent of all infestations in agricultural land.
By discovering a root-knot nematode parasitism gene that's essential for the nematode to infect crops, the scientists have developed a resistance gene effective against all four species.
Using a technique called RNA interference (RNAi), the researchers have effectively turned the nematode's biology against itself.
They genetically modified Arabidopsis, a model plant, to produce double-stranded RNA (dsRNA) to knock out the specific parasitism gene in the nematode when it feeds on the plant roots.
Concept Builder
Long double-stranded RNAs (dsRNAs; typically > 200nt) can be used to silence the expression of target genes in a variety of organisms and cell types (e.g.; worms, fruit flies and plants).
Upon introduction, the long dsRNAs enter a cellular pathway that is commonly referred to as the RNA interference (RNAi) pathway.
First, the dsRNA get processed into 20-25 nucleotide (nt) small interfering RNAs (siRNAs) by an RNase III-like enzyme called Dicer (initiation step).
Then, the complexes (RISCs), unwinding in the process.
The siRNA strands subsequently guide the RISCs to complementary RNA molecules, where they cleave and destroy the cognate RNA (effecter step).
Cleavage of cognate RNA takes place near the middle of the region bound by the siRNA strand.
In mammalian cells, introduction of long dsRNA (>30 nt) initiates a potent antiviral response, exemplified by nonspecific inhibition of protein synthesis and RNA degradation.
The mammalian antiviral response can be bypassed. however, by the introduction or expression of siRNAs.
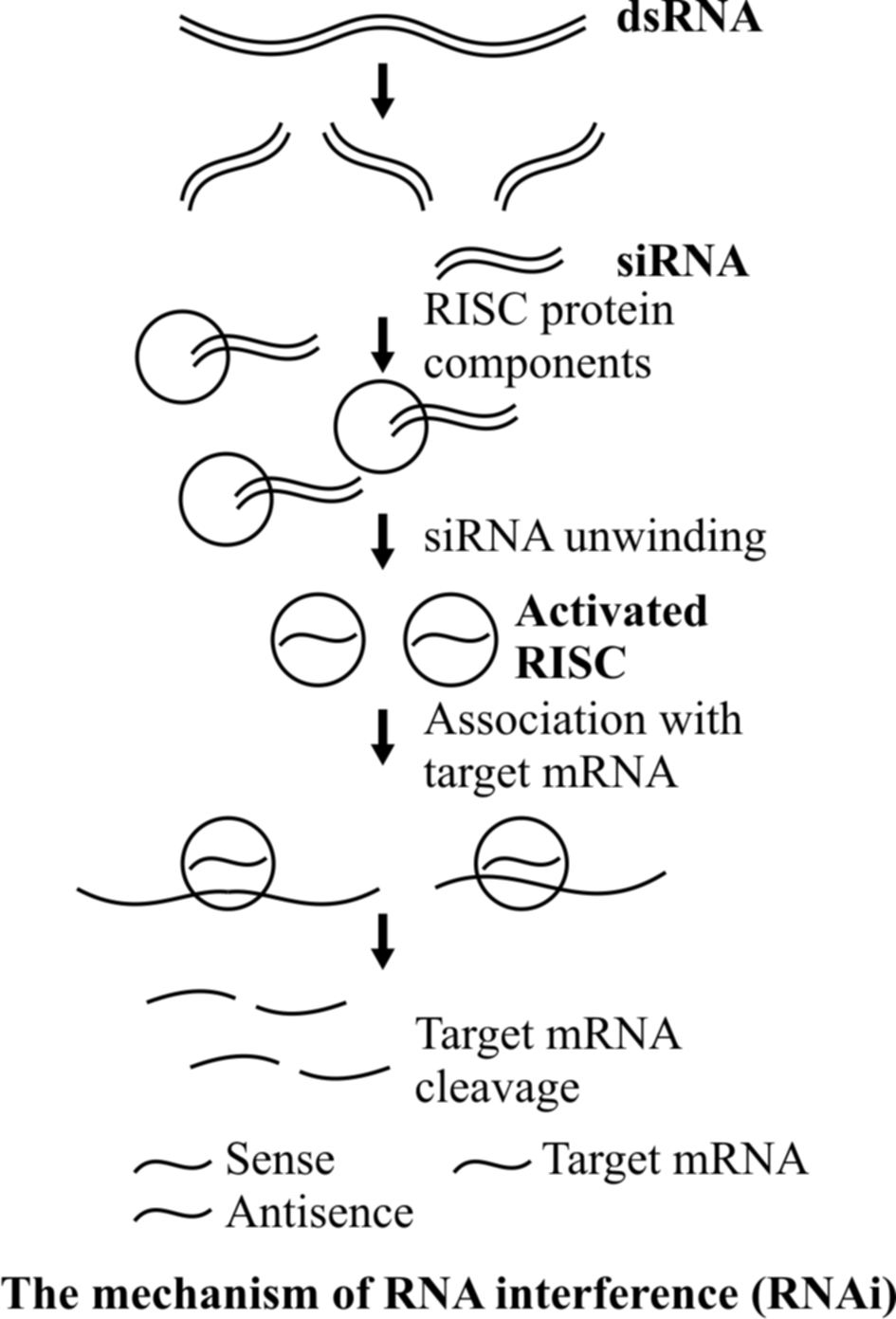
RNAi takes place in all eukaryotic organisms as a method of cellular defense.
This method involves silencing of a specific mRNA due to a complementary dsRNA molecule that binds to and prevents translation of the mRNA (silencing).
The source of this complementary RNA could be from an infection by viruses having RNA genomes or mobile genetic elements (transposons) that replicate via an RNA intermediate.
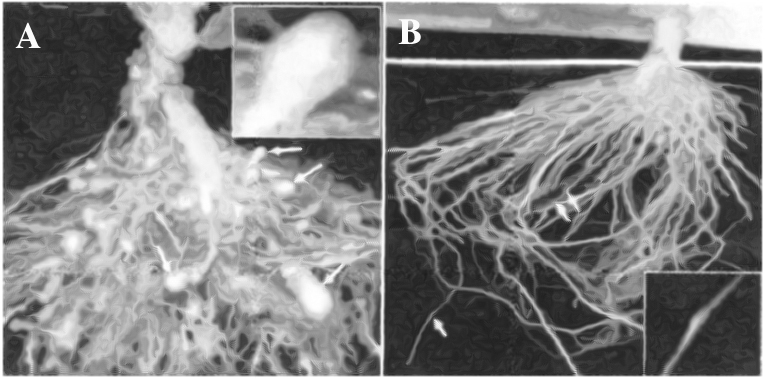
(A) Roots of a typical control plant (B) Transgenic plant roots 5 days after deliberate
infection of nematode but protected through novel mechanism.
Using Agrobacterium vectors, nematode-specific genes were introduced into the host plant.
The introduction of DNA was such that it produced both sense and anti-sense RNA in the host cells.
These two RNAs being complementary to each other formed a double stranded RNA that initiated RNAi and thus, silenced the specific mRNA of the nematode.
The consequence was that the parasite could not survive in a transgenic host expressing specific interfering RNA.
The transgenic plant therefore got itself protected from the parasite.
This knocked out the parasitism gene in the nematode and disrupted its ability to infect plants.
"No natural root-knot resistance gene has this effective range of root-knot nematode resistance."
The efforts have been directed primarily at understanding the molecular tools the nematode uses to infect plants.
This is a prerequisite for bioengineering durable resistance to these nematodes in crop plants.
Biotechnological applications in medicine
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
BIOTECHNOLOGICAL APPLICATION IN MEDICINE
1. Therapeutic agents
Proteins with potential as pharmaceutical agents are produced by using genetically engineered organisms.
Enzymes have also been used for this purpose, e.g., DNase I and alginate lyase have been used in aerosols.
Some known examples are given below:
1. Human growth hormone obtained from E. coli is used for treatment of dwarfness.
2. Chorionic gonadotropin hormone produced by genetic engineering is used for treatment of infertility.
3. Interferons produced by E. coli are commercially used for treatment of viral infections and cancer. Interferons were first obtained through DNA recombinant technique by Charles Weisman in 1980. He inserted the gene for interferon production in E. coli.
4. Interleukins produced by E. coli are used for stimulating immunity system.
5. Tissue Plasminogen Activator (TPA) -an enzyme is used for dissolving blood clot after heart attack and stroke.
6. Antihemophilic human factor VIII is used by people with hemophilia to prevent and control bleeding or to prepare them for surgery.
7. Platelet derived growth factor produced by recombinant DNA technology is useful for stimulating wound healing.
8. Penicillin G acylase is also produced by genetic engineering. This enzyme is used for converting penicillin into 6-amino penicilline acid for the formation of new antibiotics.
2. Genetically engineered insulin
Since the discovery of insulin by Banting and Best (1921), and its use for the treatment of diabetes, it was derived from pancreatic glands of abattoir animals.
This hormone, produced and secreted by the beta cells of the pancreas islets of Langerhans, regulates the use and storage of food, particularly carbohydrates.
Although bovine and porcine insulin is similar to human insulin, their composition is slightly different.
It, therefore, causes adverse effects due to regular injection, this being a foreign substance.
This observation led to the synthesis of human insulin which is chemically identical to its naturally produced component.
Insulin consists of 51 amino acids forming two short polypeptide chains-chain A having 21 amino acids and chain B with 30 amino acids.
The two chains are linked by disulfide bond. In animals, including humans, insulin occurs as proinsulin.
It is made of chain A, chain B and chain C (30 amino acids). As the insulin matures, chain C is removed.
The genetic engineering of insulin begins with identification and separation of DNA sequences coding for chain A and chain B.
This was found to be present at the top of the short arm of the eleventh chromosome.
It contains 153 nucleotides-63 nucleotides for chain A and 90 nucleotides for chain B.
These sequences were introduced into plasmid (pBR322) of Escherichia coli -common human colon bacterium.
It is said to be the factory used in genetic engineering of insulin.
In E. coli, -galactosidase controls the transcription of these genes, therefore, insulin gene needs to be tied to this enzyme.
The protein formed by E. coli consists partly of -galactosidase joined to either A or B chain of insulin.
These are then extracted from -galactosidase fragment and purified.
The two chains are mixed and reconnected in a reaction that forms disulfide bridges resulting in pure humulin-the synthetic human insulin.
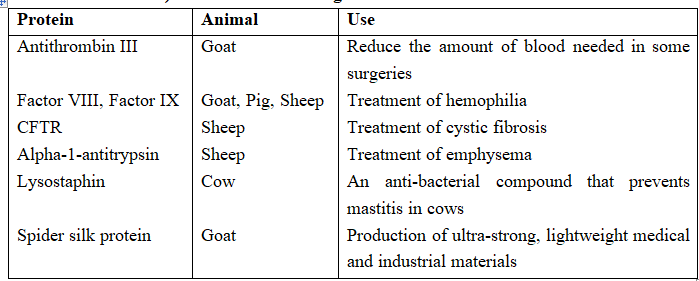
Proteins with Therapeutic and Industrial Value that have been Produced (but not Commercialized) in the Milk of Transgenic Animals
GENE THERAPY
Much attention has been focussed on the so-called genetic metabolic diseases in which a defective gene causes an enzyme to be either absent or ineffective in catalyzing a particular metabolic reaction effectively.
A potential approach to the treatment of genetic disorders in man is gene therapy.
This is a technique whereby the absent or faulty gene is replaced by a working gene, so that the body can make the correct enzyme or protein and consequently eliminate the root cause of the disease.
The first clinical gene therapy was given in 1990 to a 4-year old girl with adenosine deaminase (ADA) deficiency.
This enzyme is crucial for the immune system to function.
The disorder is caused due to the deletion of the gene for adenosine deaminase.
In some children, ADA deficiency can be cured by bone marrow transplantation; in others, it can be treated by enzyme replacement therapy, in which functional ADA is given to the patient by injection.
But the problem with both of these approaches is that they are not completely curative.
As a first step towards gene therapy, lymphocytes from the blood of the patient are grown in a culture outside the body.
A functional ADA cDNA (using a retroviral vector) is then introduced into these lymphocytes, which are subsequently returned to the patient.
However, as these cells are not immortal, the patient requires periodic infusion of such genetically engineered lymphocytes.
However, if the gene isolated from bone marrow cells producing ADA is introduced into cells at early embryonic stages, it could be a permanent cure.
Before treatment for a genetic disease can begin, an accurate diagnosis of the genetic defect needs to be made.
It is here that biotechnology is also likely to have a great impact in the near future.
Genetic engineering research has produced a powerful tool for pinpointing specific diseases rapidly and accurately.
Short pieces of DNA called DNA probes can be designed to stick very specifically to certain other pieces of DNA.
The technique relies upon the fact that complementary pieces of DNA stick together.
DNA probes are more specific and have the potential to be more sensitive than conventional diagnostic methods, and it should be possible in the near future to distinguish between defective genes and their normal counterparts, an important development.
Molecular Diagnosis
For effective treatment of a disease, early diagnosis and understanding its pathophysiology is very important.
Using conventional methods of diagnosis (serum and urine analysis, etc.), early detection is not possible.
Recombinant DNA Technology, Polymerase Chain Reaction (PCR) and Enzyme Linked Immuno-Sorbent Assay (ELISA) are some of the techniques that serve the purpose of early diagnosis.
Presence of a pathogen (bacteria, viruses, etc.) is normally suspected only when the pathogen has produced a disease symptom.
By this time the concentration of pathogen is already very high in the body.
However, very low concentration of a bacteria or virus (at a time when the symptoms of the disease are not yet visible) can be detected by amplification of their nucleic acid by PCR, which is now routinely used to detect HIV in suspected AIDS patients.
It is being used to detect mutations in genes in suspected cancer patients too.
It is a powerful technique to identify many other genetic disorders.
DNA is usually isolated from White blood cells & has to be cut into smaller pieces to be analysed.
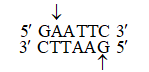
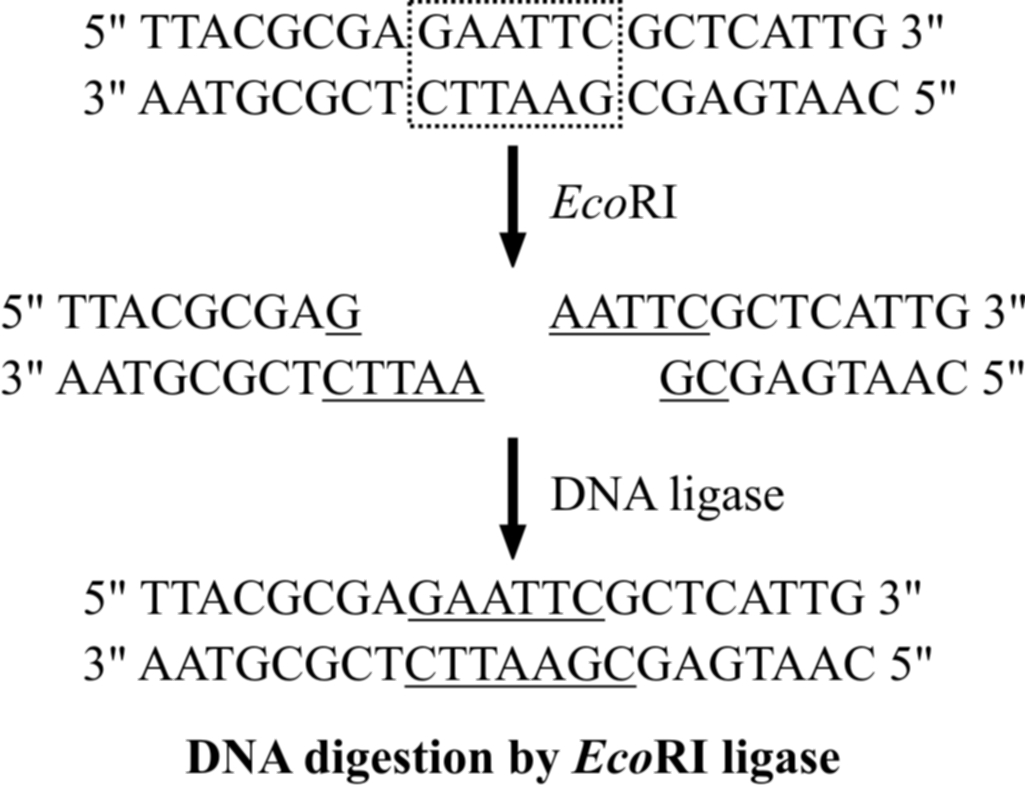
This is accomplished by restriction enzymes. Eco RI (a restriction enzyme from E. coli) will cut DNA wherever the sequence GAATTC appears.
Exposure to this enzyme results in the DNA being chopped into millions of fragments of varying size, called restriction fragments.
Once cut, the DNA is loaded into a well on one end of a slab of gel.
The fragments are then separated according to size by electrophoresis.
As electric current passes through the gel, the fragments move according to size.
The bigger fragments stay close to the origin, and the smaller fragments move farther down the length of the gel.
The DNA is then denatured (by exposure to alkaline solutions) to render the DNA single stranded (instead of the natural double-stranded form).
Since the gel is difficult to handle, the DNA is transferred to a nitro cellulose paper to create a Southern blot (named after the researcher who developed the procedure).
The DNA probe which is radioactively labeled (or fluorescently labeled) is then applied to the Southern blot.
Since the probe is also single-stranded, it will seek the single-stranded DNA fragments that are complementary, and undergo hybridization.
The excess probe is washed out and only the bound probe will remain on the Southern blot paper.
This is then laid on an X-ray film.
The radioactive probe will leave bands on the X-ray film.
Depending on the type of probe used, there could be hundreds of bands (much like bar codes) or only a few bands present on the X-ray film.
By having several wells on the end of the gel, several samples can be loaded, and DNA fragments in corresponding lanes can be analyzed concurrently.
By running control samples, with known DNA fragment sizes, on the same gel with patient samples, it is possible to identify changes in the size of a DNA fragment and, therefore, a change in a specific gene.
Since each step takes about a day and since samples are batched, the procedure ordinarily takes one to two weeks to complete.
ELISA is based on the principle of antigen-antibody interaction. Infection by pathogen can be detected by the presence of antigens (proteins, glycoproteins, etc.) or by detecting the antibodies synthesised against the pathogen.
Transgenic Animals
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
Transgenic Animal
There are various definitions for the term transgenic animal.
A transgenic animal is one whose genome has been changed to carry genes from other species.
The nucleus of all cells in every living organism contains genes made up of DNA.
These genes store information that regulates how our bodies form and function.
Genes can be altered artificially, so that some characteristics of an animal are changed.
For example, an embryo can have an extra, functioning gene from another source artificially introduced into it, or a gene introduced which can knock out the functioning of another particular gene in the embryo.
Animals that have their DNA manipulated in this way are known as transgenic animals.
The majority of transgenic animals produced so far are mice, the animal that pioneered the technology.
The first successful transgenic animal was a mouse. A few years later, it was followed by rabbits, pigs, sheep, and cattle.
How are transgenic animals produced?
To date, there are three basic methods of producing transgenic animals:
(i) DNA microinjection
(ii) Retrovirus-mediated gene transfer
(iii) Embryonic stem cell-mediated gene transfer
Gene transfer by microinjection is the predominant method used to produce transgenic farm animals.
Since the insertion of DNA results in a random process, transgenic animals are mated to ensure that their offsprings acquire the desired transgene.
However, the success rate of producing transgenic animals individually by these methods is very low and it may be more efficient to use cloning techniques to increase their numbers.
For example, gene transfer studies revealed that only 0.6% of transgenic pigs were born with a desired gene after 7,000 eggs were injected with a specific transgene.
How do transgenic animals contribute to human welfare?
The benefits of these animals to human welfare can be grouped into following areas:
(1) Agriculture (2) Medicine (3) Industry
The examples below are not intended to be complete but only to provide a sampling of the benefits.
1. Agricultural Applications
(a) Breeding: Farmers have always used selective breeding to produce animals that exhibit desired traits (e.g., increased milk production, high growth rate). Traditional breeding is a time-consuming, difficult task. When technology using molecular biology was developed, it became possible to develop traits in animals in a shorter time and with more precision. In addition, it offers the farmer an easy way to increase yields.
(b) Quality: Transgenic cows exist that produce more milk or milk with less lactose or cholesterol, pigs and cattle that have more meat on them, and sheep that grow more wool. In the past, farmers used growth hormones to spur the development of animals but this technique was problematic, especially since residue of the hormones remained in the animal product.
(c) Disease resistance : Scientists are attempting to produce disease-resistant animals, such as influenza-resistant pigs, but a very limited number of genes are currently known to be responsible for resistance to diseases in farm animals.
2. Medical Applications
(a) Xenotransplantation : Patients die every year for lack of a replacement heart, liver, or kidney. For example, about 5,000 organs are needed each year in the United Kingdom alone. Transgenic pigs may provide the transplant organs needed to alleviate the shortfall. Currently, xenotransplantation is hampered by a pig protein that can cause donor rejection but research is underway to remove the pig protein and replace it with a human protein.
(b) Nutritional supplements and pharmaceuticals: Products such as insulin, growth hormone, and blood anti-clotting factors may soon be or have already been obtained from the milk of transgenic cows, sheep, or goats. Research is also underway to manufacture milk through transgenics for treatment of debilitating diseases such as phenylketonuria (PKU), hereditary emphysema, and cystic fibrosis.
In 1997, the first transgenic cow, Rosie, produced human protein-enriched milk at 2.4 grams per litre. This transgenic milk is a more nutritionally balanced product than natural bovine milk and could be given to babies or the elderly with special nutritional or digestive needs. Rosie's milk contains the human gene -lactalbumin.
(c) Vaccine safety: Transgenic mice are being developed for use in testing the safety of vaccines before they are used on humans. Transgenic mice are being used to test the safety of the polio vaccine. If successful and found to be reliable, they could replace the use of monkeys to test the safety of batches of the vaccine.
3. Industrial Applications
In 2001, two scientists at Nexia Biotechnologies in Canada spliced spider genes into the cells of lactating goats.
The goats began to manufacture silk along with their milk and secrete tiny silk strands from their body by the bucketful.
By extracting polymer strands from the milk and weaving them into thread, the scientists can create a light, tough, flexible material that could be used in such applications as military uniforms, medical microsutures, and tennis racket strings.
Toxicity-sensitive transgenic animals have been produced for chemical safety testing.
Microorganisms have been engineered to produce a wide variety of proteins, which in turn can produce enzymes that can speed up industrial chemical reactions.
The anthrax bacterium sent through letters after September 2001.
Mass-produced pathogens or their toxins are delivered either as powder or in the form of spray, using a variety of delivery devices.
Bioweapons (a) are low-cost weapons, (b) cause for more casualities than chemical or conventional weapons, and (c) bioweapon agents are invisible, and extremely difficult to detect.
These features make bioweapon agents very convenient for use by terrorists and even governments, and both have used them on a limited scale.
The possible defences against bioweapons include the use of respirator or gas mask, vaccination, administration of appropriate antibiotics, and decontamination. In addition, sensitive detection systems should be develped to control and minimise damage.
Ethical Issues
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
BIOETHICS
Ethics includes a set of standards by which a community regulates its behaviour and decides as to which activity is legitimate and which is not.
Therefore, bioethics may be viewed as a set of standards that may be used to regulate our activities in relation to the biological world.
Biotechnology, particularly recombinant DNA technology, is focussed on' exploiting the biological world in ways that are usually unprecedented.
Therefore, biotechnology has been labelled variously, ranging from 'unnatural' to 'detremental, to 'biodiversity'.
The major bioethical concerns pertaining to biotechnology are summarised below:
(i) Use of animals in biotechnology causes great suffering to them.
(ii) When animals are used for production of pharmaceutical proteins, they are virtually reduced to the status of a 'factory'.
(iii) Introduction of a transgene from one species into another species violates the 'integrity of species'.
(iv) Transfer of human genes into animals (and vice-versa ) dilutes the concept of 'humanness'.
(v) Biotechnology is disrespectful to living beings, and only exploits them for the benefit of human beings.
(vi) Biotechnology may pose unforeseen risks to the environment, including risk to biodiversity.
These arguments may seem quite attractive.
It may be pointed out that biotechnology usually does only what was being done before.
However, biotechnologies do these things on a much larger scale and at a much faster rate.
Each society has to evaluate for itself the validity of these and other arguments related to biotechnology.
It also has to decide the kinds of activities that it considers acceptable, and those that it does not.
Going beyond the morality of such issues, the biological Significance of such things is also important.
Genetic modification of organisms can have unpredictable results when such organisms are introduced into the ecosystem.
Therefore, the Indian Government has set up organisations such as GEAC (Genetic Engineering Approval Committee), which will make decisions regarding the validity of GM research and the safety of introducing GM-organisms for public services.
The modification/usage of living organisms for public services (as food and medicine sources, for example) has also created problems with patents granted for the same.
BIOPATENT
A patent is the right granted by a government to an inventor to prevent others from commercial use of his invention.
A patent is granted for (a) an invention (including a product), (b) an improvement in an earlier invention, (c) the process of generating a product, and (d) a concept or design.
Initially, patents were granted for industrial inventions, etc.
But at present, patents are being granted for biological entities and for products derived from them ; these patents are called biopatents.
Primarily, industrialised countries, like U.S.A., Japan and members of European Union, are awarding Biopatents.
Biopatents are awarded for the following : (i) strains of microorganisms, (ii) cell lines, (iii) genetically modified strains of plants and animals, (iv) DNA sequences, (v) the proteins encoded by DNA sequences, (vi) various biotechnological procedures, (vii) production processes, (viii) products, and (ix) product applications.
There has been a great deal of opposition from various social groups to the patenting of life forms.
The nature of these objections is mainly ethical and political.
The arguments in favour of biopatents are primarily of increased economic growth.
Concept Builder
Intellectual property (IP) is a term referring to a number of distinct types of creation of the mind for which a set of exclusive rights are recognized and the corresponding fields of law.
Under intellectual property law, owners are granted certain exclusive rights to a variety of intangible assets, such as musical, literary, and artistic works; discoveries and inventions; and words, phrases, symbols and designs.
Common types of intellectual property include copyrights, trademarks, patents, industrial design rights and trade secrets in some jurisdictions.
Many biotechnology patents are very broad in their coverage.
For example, one patent covers 'all transgenic plants of Brassica family'.
Such broad patents are considered .morally unacceptable and fundamentally inequitable, since these would enable financially powerful corporations to acquire monopoly control over biotechnological processes.
They may, in the end, even come to control the direction of agricultural research, including plant breeding.
Such a position would pose a threat to the global food security.
Any organisations and multinational companies exploit and/or patent biological resources or bioresources of other nations without proper authorisation from the countries concerned; this is known as biopiracy.
The industrialised nations are rich in technology and financial resources but poor in biodiversity and traditional knowledge related to the utilisation of the bioresources.
In contrast, developing nations are poor in technology and financial resources, but are rich in biodiversity and traditional knowledge related to bioresources.
Biological resources or bioresources include all those organisms that can be used to derive commercial benefits.
Traditional knowledge related to bioresources is the knowledge developed by various communities over long periods of history, regarding the utilisation of the bioresources, e.g., use of herbs, etc. as drugs.
Often, this traditional knowledge can be exploited to develop modern commercial processes.
The traditional knowledge suggests the direction to be followed, and saves considerable time, effort and expenditure for their commercialisation.
Institutions and companies of industrialised nations are collecting and exploiting the bioresources, as follows,
(i) They are collecting and patenting the genetic resources themselves. For example, a patent granted in U.S.A covers the entire 'basmati' rice germplasm indigenous to our country.
(ii) The bioresources are being analysed for identification of valuable biomolecules. A biomolecule is a compound produced by a living organism. The biomolecules are then patented and used for commercial activities.
(iii) Useful genes are isolated from the bioresources and patented. These genes are then used to generate commercial products.
(iv) The traditional knowledge related to bioresources is utilised to achieve the above objectives. In some cases, the traditional knowledge itself may be the subject of a patent.
A west African plant, Pentadiplandra brazzeana produces a protein called brazzein, which is approximately 2,000 times as sweet as sugar.
In addition, brazzein is a low-calorie sweetener.
Local people have known and used the super-sweet berries of this plant for centuries.
But the protein brazzein was patented in U.S.A.
Subsequently, the gene encoding brazzein was also isolated, sequenced and patented in U.S.A.
It is proposed to transfer the brazzein gene into maize and express it in maize kernels.
These kernels will then be used for the extraction of brazzein.
This development could have serious implications for countries exporting large quantities of sugar.
