 ACME SMART PUBLICATION
ACME SMART PUBLICATION
principle of biotechnology
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
PRINCIPLES OF BIOTECHNOLOGY
Biotechnology can be broadly defined as "using living organisms or their products for commercial purposes".
As such, biotechnology has been practiced by human society since the beginning of recorded history in such activities as baking bread, brewing alcoholic beverages, or breeding food crops or domestic animals.
A narrower and more specific definition of biotechnology is "the commercial application of living organisms or their products, which involves the deliberate manipulation of their DNA molecules".
This definition implies a set of laboratory techniques developed within the last 20 years that have been responsible for the tremendous scientific and commercial interest in biotechnology.
Some other available definitions of biotechnology are :
''The application of biological organism, system or processes to manufacturing & service industries".
-British Biotechnologist
"The integrated use of biochemistry, microbiology & genetic engineering sciences in order to achieve technological (industrial) application of capabilities of micro-organisms, cultured tissue cells & parts there of".
-European Federation of Biotechnology
"The controlled use of biological agents, such as micro-organisms or cellular components, for beneficial use ".
-US National Science Foundation
Development of biotechnology may be studied considering its growth that occurred in two phases : (1) The traditional (old) biotechnology, and (2) The new (modern) biotechnology.
1. Traditional Biotechnology:
It includes the processes that are based on the natural capabilities of microorganisms.
The traditional biotechnology is also called conventional technology which has been used for many centuries.
Curd, vinegar, ghee, wine and beer and other alcoholic beverages, idli, dosa, cheese, paneer and some other foods have been produced using traditional biotechnology.
In Indian Ayurveda, production of 'Asva', 'Arista', etc. is done through traditional biotechnology.
According to some people, traditional biotechnology, is therefore, called an art rather than a science.
2. Modern Biotechnology:
When highly new and useful traits in crop varieties and animal breeds are created with the help of genetic engineering, it is called modern biotechnology.
It was developed during 1970.
For example, in vitro fertilization leading to a 'test tube baby', synthesizing a gene and using it, developing a DNA vaccine or correcting a defective gene are all a part of modern biotechnology.
Among many, the two main techniques that gave birth to modern biotechnology are as follows:
(i) Genetic Engineering: The techniques which change the chemistry of genetic material (DNA and RNA) to introduce these into host organisms and thus alter the phenotype of the host organism are called genetic engineering (= "Recombinant DNA Technology").
(ii) To maintain the microbial contamination free (Sterile) surrounding in chemical engineering: Due to such type of maintenance only desired microorganisms/cells will be formed in large number for manufacture of biotechnological products such as antibiotics, vaccines, enzymes, hormones, blood clotting factors, etc. It is essential to have complete aseptic conditions.
Concept of Genetic Engineering
Combining DNA from different existing organisms like plants, animals, bacteria etc. results in modified organism with a combination of trait from the parents.
This sharing of DNA information occurs naturally through sexual reproduction and has been exploited in plant and animal breeding for number of years.
However sexual reproduction (recombination) can occur between individuals of same species.
Genetic engineering is the manipulation of prokaryotic as well as eukaryotic DNA.
It involves breakage of a DNA molecule at two desired places to isolate a specific DNA segment and then insert it in another DNA molecule at a desired position.
The product thus obtained is called recombinant DNA and the technique often called genetic engineering.
The cutting of the DNA at specific location became possible by so-called 'molecular scissors', i.e., restriction enzymes.
In chromosomes, there is specific 'ori' site or origin of replication which initiates the replication.
Therefore, for duplication of any foreign DNA in an organism, it should be linked with the 'ori' sites so that the foreign DNA can duplicate and multiply within the organism.
This is called gene cloning and can produce multiple copies of any template DNA.
The construction of the first recombinant DNA emerged from the possibility of linking a gene encoding antibiotic resistance with a native plasmid (autonomously replicating circular extra-chromosomal DNA) of Salmonella typhimurium.
Stanley Cohen and Herbert Boyer accomplished this in 1972 by isolating the antibiotic resistance gene by cutting out a piece of DNA from a plasmid which was responsible for conferring antibiotic resistance.
These plasmid DNA molecules act as vectors to transfer the piece of DNA attached to it.
As mosquito acts as an insect vector to transfer the malarial parasite into human body.
In the same way, a plasmid can be used as vector to deliver an alien piece of DNA into the host organism.
The linking of antibiotic resistance gene with the plasmid vector became possible with the enzyme DNA ligase, which acts on cut DNA molecules and joins their ends.
This makes a new combination of circular autonomously replicating DNA created in vitro and is known as recombinant DNA.
When this DNA is transferred into Escherichia coli, a bacterium closely related to Salmonella, it could replicate using the new host's DNA polymerase enzyme and make multiple copies.
The ability to multiply copies of antibiotic resistance gene in E. coli was called cloning of antibiotic resistance gene in E. coli.
Therefore, there are three basic steps in genetically modifying an organism :
(i) Identification of DNA with desirable genes;
(ii) Introduction of the identified DNA into the host;
(iii) Maintenance of introduced DNA in the host and transfer of the DNA to its progeny .
tools of recombinant DNA technology(RDT)
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
TOOLS OF RECOMBINANT DNA TECHNOLOGY
The technology or genetic engineering involves restriction enzymes, ligase enzymes, polymerase enzymes, vectors and the host organism.
Restriction Enzymes
In the late 1960's, scientists Stewart Linn and Werner Arber isolated samples of the two types of enzymes responsible for phage growth restriction in Escherichia coli (E. coli) bacteria.
One of these enzymes methylated DNA, while the other cleaved unmethylated DNA at a wide variety of locations along the length of the molecule. The first type of enzyme was called a "methylase" while the other was called a "restriction nuclease".
These enzymatic tools were important to scientists who were gathering the tools needed to "cut and paste" DNA molecules.
What was needed now was a tool that would cut DNA at specific sites, rather than at random sites along the length of the molecule, so that scientists could cut DNA-molecules in a predictable and reproducible way.
Site-specific Nuclease
This important development came when H.O. Smith, K.W. Wilcox, and T.J. Kelley isolated and characterized the first restriction nuclease whose functioning depended on a specific DNA nucleotide sequence.
Working, with Haemophilus influenzae bacteria, this group isolated an enzyme, called Hind II, that always cut DNA molecules at a particular point within a specific sequence of six base pair.
This sequence is :
5' G T (pyrimidine : Tor C) (purine : A or G) A C 3'
3' C A (purine : A or G) (pyrimidine : T or C) T G 5'
They found that the Hind II enzyme always cuts directly in the center of this sequence.
Wherever this particular sequence of six base pairs occurs unmodified in a DNA molecule, Hind II will cleave both DNA strands or backbones between the 3rd and 4th base pairs of the sequence.
Moreover, Hind II will only cleave a DNA molecule at this particular site. For this reason, this specific base sequence is known as the "recognition sequence" for Hind II.
Hind II is just one example of the class of enzymes known as restriction nucleases.
In fact, more than 900 restriction enzymes, some sequences specific and some not, have been isolated from over 230 strains of bacteria since the initial discovery of Hind II.
These restriction enzymes generally have names that reflect their origin.
The first letter (in italics) of the name comes from the genus and the second two letters (in italics) come from the species of the prokaryotic cell from which they were isolated.
Next is the strain of the organism and last is the roman numeral indicating the order of discovery.
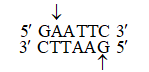
For example, EcoRI comes from Escherichia coli RY strain and was the first endonuclease isolated from bacteria, while Hind II comes from Haemophilus influenzae strain Rd. Numbers following the nuclease names indicate the order in which the enzymes were isolated from single strains of bacteria.
Nucleases are further described by addition of the prefix "endo" or "exo" to the name: The term "endonuclease" applies to sequence specific nucleases that break nucleic acid chains somewhere in the interior, rather than at the ends of the molecule.
Nuclease that function by removing nucleotides from the ends of the molecules are called "exonucleases".
Three main classes of restriction endonucleases – type-I, type-II and type-III have been described, each distinguished by a slightly different mode of action.
Out of these three types, type-I and type-III restriction enzymes are not used in recombinant DNA technology.
Type-II restriction enzymes are used in recombinant DNA technology, because they can be used In vitro to recognise and cut within the specific DNA sequence typically consisting of 4-8 nucleotide.
Type-I enzymes recognize specific sites within the DNA but do not cut at these sites.
Hence heterogeneous population of DNA fragments is produced, and therefore, type I enzymes do not take part in the technology. Type-III enzymes recognise a specific sequence of DNA molecule. Thus, products of type-III enzymes are homogeneous population of DNA fragments, so they cannot be used for genetic engineering experiments.
Table-I : Recognition Sequences of Several Restriction Endonucleases
(arrow indicates the site of cleavage)

The DNA segments cut by restriction enzymes are palindromic i.e., the nucleotide sequence of these DNA pieces read the same both, backwards and forward when orientation of reading is kept same, e.g., madam.
Blunt or flush ends are produced by many restriction enzymes which cleave both stands of DNA at exactly the same nucleotide position, in the centre of recognition site. For example, Sma I recognises 6 nucleotide palindromic sequence.
5'-C-C-C-G-G-G-3'
3'-G-G-G-C-C-C-5'
It cuts both DNA strands producing blunt ends.
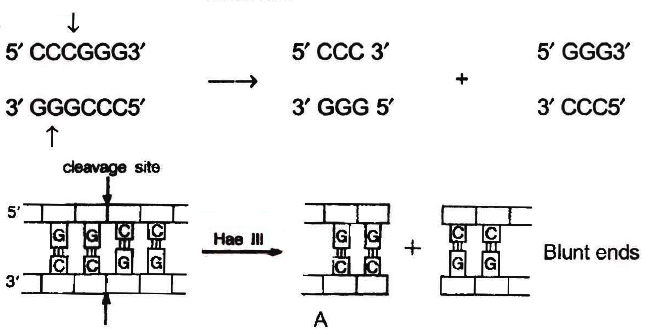
Action of restriction enzymes : The nucleotide sequences recognised and
cut by widely used restriction endonucleases A. Hae III
Sticky or cohesive ends are produced when restriction enzymes do not cut DNA at the same nucleotide position but cut the recognition sequence unequally. This produces short, single-stranded overhangs at each end. These are known as sticky ends. For example, Eco RI recognises 6 nucleotide palindromic sequence.
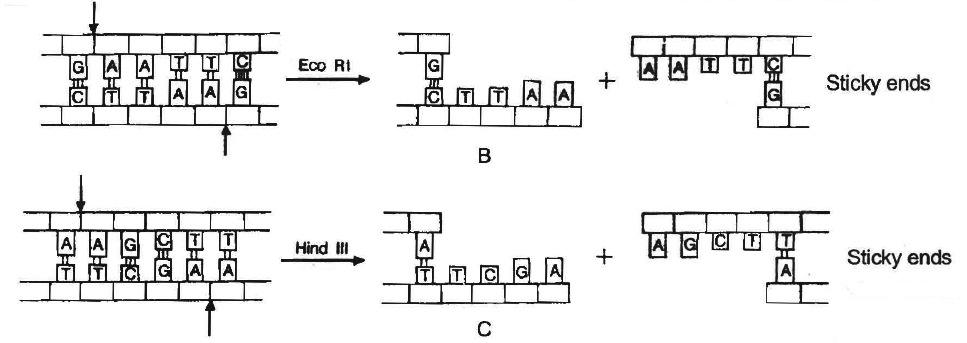
Action of restriction enzymes : The nucleotide sequences recognised and
cut by widely used restriction endonucleases B. Eco RI, C. Hind IIIC
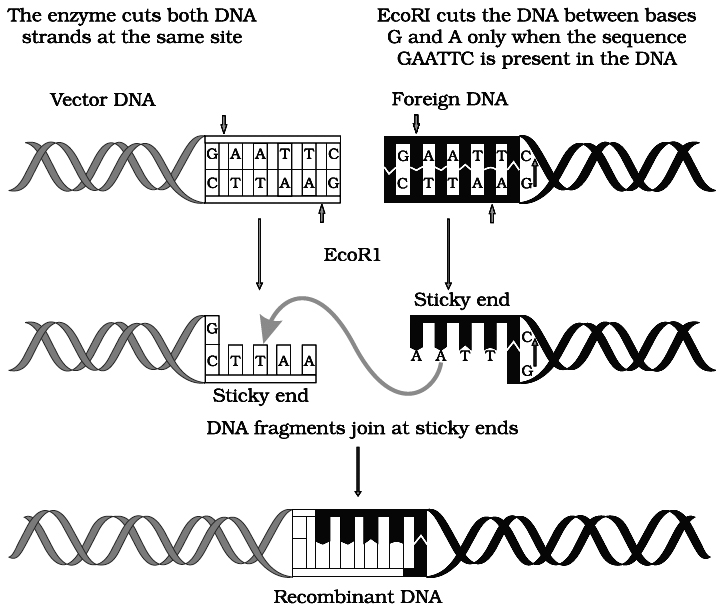
Action of restriction enzyme Eco RI and formation of recombinant DNA.
 This restriction endonuclease cuts both DNA strands unequally, producing 5' overhangs of 4 nucleotides.
This restriction endonuclease cuts both DNA strands unequally, producing 5' overhangs of 4 nucleotides.
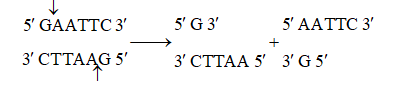
The stickiness helps enzyme ligase to make the DNA pieces join.
Other Enzymes used in Recombinant DNA Technology
In addition to restriction enzymes, there are several other enzymes that play an important role in DNA technology.
Three of the important ones are DNA ligase, alkaline phosphatase and DNA polymerase.
(a) DNA Ligase : This enzyme forms phosphodiester bonds between adjacent nucleotides and covalently links two individual fragments of double-stranded DNA. The action of the ligase enzyme requires a phosphate group at the 5' carbon of one nucleotide and a hydroxyl group at the 3' carbon of the adjacent nucleotide to form the phosphodiester bond between these two nucleotides. The enzyme used most often in the rDNA technology is T4 DNA ligase, which is encoded by phage T4.
(b) Alkaline Phosphatase (AP) : As mentioned above, ligation absolutely requires the presence of 5' phosphate group at the DNA site to be ligated. If this phosphate group is removed, this DNA cannot be ligated. The enzyme alkaline phosphatase is used to remove the phosphate group from the 5' end of a DNA molecule, leaving a free 5' hydroxyl group. This enzyme can be isolated from bacteria (BAP) or calf intestine (CAP). It is used to prevent unwanted self-ligation of vector DNA molecules in procedures of rDNA technology. However, ligation of the vector to the insert can occur as the insert still has its 5' phosphate.
(c) DNA Polymerase : DNA Pol I enzyme polymerizes the DNA synthesis on DNA template or complementary DNA (cDNA). It also catalyses a 5' 3' and 3' 5' exonucleolytic degradation of DNA. The other two enzymes are DNA polymerase II (DNA pol II) and DNA polymerase III (DNA pol III). These have almost similar catalytic activity. DNA pol III is about several times more active than the other two. Where there is preformed DNA template, it produces a parallel strand in the presence of ATP.
Separation and Isolation of DNA Fragments
After the cutting of DNA by restriction enzymes, fragments of DNA are formed.
These fragments can be separated by a technique called gel electrophoresis.
Electrophoresis is a technique of separation of charged molecules under the influence of an electrical field so that they migrate in the direction of electrode bearing the opposite charge, viz., positively charged molecules move towards cathode (-ve electrode) and negatively charged molecules travel towards anode (+ve electrode) through a medium/matrix.
This technique was developed by A. Tiselius in 1937.
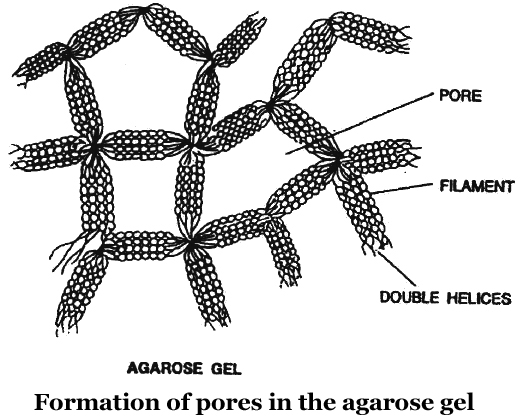
Now a days, the most commonly used matrix is agarose which is a polysaccharide extracted from sea weeds.
DNA fragments separate according to size through the pores of agarose gel.
Hence, the smaller the fragment size, the farther it moves.
Agarose dissolve in hot water, when this solution is cooled, double helices form and become arranged laterally and produce thick filaments.
These filaments become cross-linked to form the gel.
Pore size depends on agarose concentration.
The separated DNA fragments can be seen only after staining the DNA with a compound known as ethidium bromide followed by exposure to UV radiation as bright orange coloured bands. The separated bands of DNA are cut out from the agarose gel and extracted from the gel piece. This step is called as elution. Several techniques are used for eluting the DNA from the gel piece. These purified DNA fragments are used in the formation of recombinant DNA by linking them with cloning vectors.
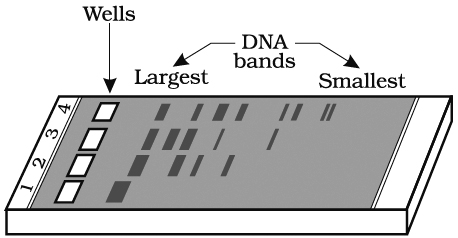
and digested set of DNA fragments (lane 2 to 4)
Cloning Vectors : Another important tool for genetic engineering is the vehicle for cloning, called vector. The vector carries a foreign DNA sequence into a given host cell. Bacterial plasmids and bacteriophages are considered the most useful. This is because :
(1) These are independent of the control of chromosomal DNA,
(2) Bacteriophage genomes occur in very large numbers in bacterial cells.
(3) The copies of plasmids per cell range from only a few to hundred or even more.
Certain essential features should be present in a DNA molecule to act as a cloning vector.
1. Origin of replication (ori): This is a DNA sequence which serves as a starting point for replication. When a DNA fragment gets associated with ori, foreign DNA into the vector would also replicate inside the host cell. Some vectors possess origin which favours formation of high copy numbers and hence preferred.
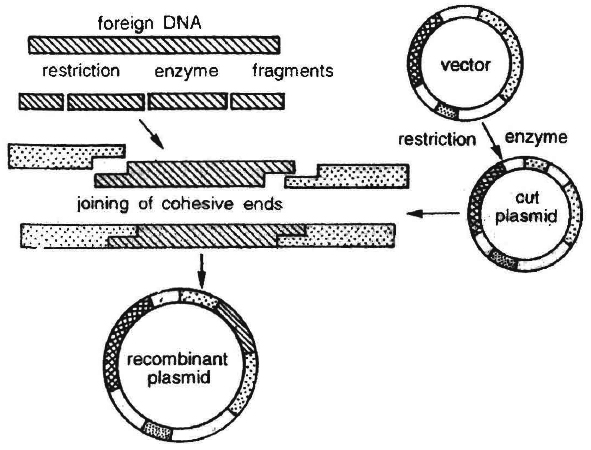
2. Selectable marker: Vector should also include a selectable marker. This is a gene which would permit the selection of host cells containing vector from amongst those which do not possess vector. Common selectable markers include genes encoding antibiotic resistance such as ampicillin resistance or enzymes such as -galactosidase (product of lac Z gene of lac operon). These genes can be identified by a colour reaction.
3. Recognition sites : Vector should possess an unique restriction site that would allow particular enzyme to cut the vector only once. This site would be recognised by the commonly used restriction enzymes. If there are more than one recognition site in the vector, several fragments would be produced. Generally the vectors used possess unique recognition sites for several restriction enzymes in a small region of DNA. This is known as polylinker or multiple cloning site (MCS). Such a cloning site offers choice of restriction enzyme.
Unique restriction endonuclease recognition site enables insertion of foreign DNA into the vector for production of recombinant DNA. The foreign DNA is inserted and made to join (ligate) at a specific restriction site generally in antibiotic resistance gene.
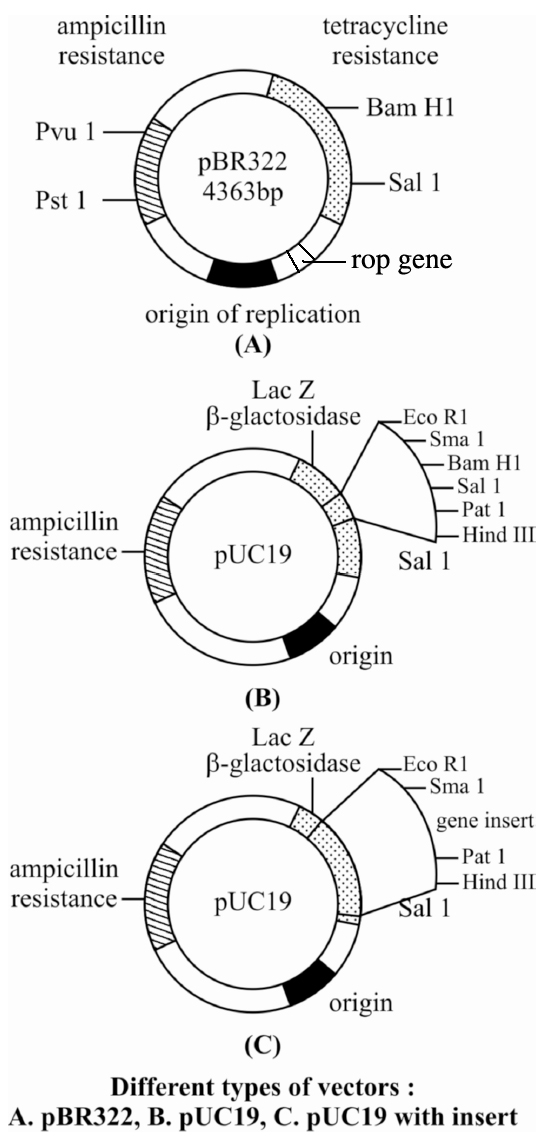
pBR322 has genes for resistance against two antibiotics tetracycline and ampicillin, an origin of replication and a variety of restriction sites for cloning of restriction fragments obtained through cleavage with a specific enzyme. Foreign DNA is inserted at a site located in one of the two genes for resistance against antibiotics, so that it will inactivate one of the two resistance genes. The insert bearing plasmid can be selected by their ability to grow in a medium containing only one of the two antibiotics and their failure to grow in a medium containing both antibiotics. The plasmids carrying no insert on the other hand, will be able to grow in a media containing one or both the antibiotics. In this way, the presence of resistance genes against ampicillin and tetracycline allow selection of Escherichia coli colonies transformed with plasmids carrying the desired foreign cloned DNA fragment.
4. Size of the vector : Cloning vector should be small in size. Large molecules have a tendency to breakdown during purification. These are also difficult to manipulate.
DIFFERENT TYPES OF VECTOR
Several types of vectors satisfying the above characters have been developed. The following are some of the commonly used vectors.
1. Plasmids:
These are extra-chromosomal, non-essential self-replicating, usually circular and double stranded DNA molecules occurring in some bacteria and also a few yeasts.
Some of the characters carried by plasmid may not be required for normal bacterial metabolism but may be of great advantage e.g. antibiotic resistance.
pBR322 is one of the standard cloning vectors widely used in gene cloning experiments.
This vector has been restructured by inserting genes for antibiotic resistance.
It is named after Boliver and Rodriguez who prepared this vector, pUC (named after University of California) is another such reconstructed plasmid vector.
The vectors mentioned above are able to replicate only in E. coli.
Therefore, many vectors constructed for eukaryotic cells are also functional in E. coli.
These vectors are called shuttle vectors.
The vectors contain two types of origin of replication and selectable marker genes-one for the eukaryotic cell and another for E. coli.
The common example of this type is yeast episomal plasmid (YEp).
In plants, Ti plasmid of bacterium Agrobacterium tumfaciens has been modified to function as vector.
2. Vectors based on bacteriophages :
Bacteriophages are viruses which infect bacterial cells produce new phages inside the host bacterium, and are released from the host cell to again infect other bacterial cells. M 13 and lambda () phages are in common use.
3. Cosmids:
These combine some features of plasmid and 'cos' (cohesive end sites) of phage lambda (cosmid = cos + plasmid).
4. YAC vectors:
Yeast artificial chromosome contain telomeric sequence, the centromere and autonomously replicating sequence from yeast chromosomes. These also have suitable restriction enzyme sites and genes useful as selectable markers.
5. BAC vectors:
Bacterial artificial chromosome is based on F plasmid (fertility) of E. coli. It contains genes for replication and maintenance of F-factor, selectable marker and cloning sites.
Colour Reaction : Due to inactivation of antibiotics, selection of recombinants becomes burdensome process because it requires simultaneous plating on two plates having different antibiotics. Thus, alternative selectable marker is developed to differentiate recombinants and non-recombinants on the basis of their ability to produce colour in the presence of a chromogenic substance. Now a recombinant DNA is inserted in the coding sequence of an enzyme -galactosidase. This causes inactivation of the enzyme which is called insertional inactivation. If the plasmid in the bacterium does not have an insert, the presence of a chromogenic substrate gives blue coloured colonies. Presence of insert results into insertional inactivation of the -galactosidase and, therefore, the colonies do not produce any colour, these colonies are marked as recombinant colonies.
6. Vectors for Cloning Genes in Plants and Animals:
We know the procedure of transferring genes into plants and animals from bacteria and viruses.
It is also known how to transfer genes to transform eukaryotic cells and force them to do what the bacteria or viruses require.
For example, Agrobacterium tumifaciens, a pathogen (disease causing agent) of several dicot plants is able to transfer a piece of DNA known as 'T-DNA' to convert normal plant cells into tumour and direct these tumour cells to secrete the chemicals required by the pathogen.
Similarly, retroviruses (cause leukosis or sarcoma types of cancer) in animals including humans are able to change normal cells into cancerous cells.
The tumour inducing (Ti) plasmid of Agrobacterium tumifaciens has been modified into cloning vector which is not pathogenic to the plants, however, it is still able to use the procedure to deliver genes of our interest into various plants.
Similarly retroviruses are used to carry desirable genes into animal cells.
Thus once a gene or DNA fragment is joined to a suitable vector it is transferred into a bacterial plant or animal host where it undergoes multiplication.
HOST CELL
Competent host cell is required for transformation with recombinant DNA.
After formation of recombinant DNA, propagation of it must occur inside a living system or a host.
Different type of available host cells are like E. coli, yeast, animal and plant cells.
The type of host cell to be used depends on the aim of cloning experiment.
Eukaryotic cells will be the preferred host for expression of some eukaryotic proteins.
Yeast cells are preferred because these are simplest eukaryotic organisms and like bacteria are single celled, genetically well characterized, easy to grow and manipulate.
Plant and animal cells can be used for protein expression either in tissue culture or as cells in the whole organism to create genetically modified (GM) crops and animals.
As DNA is hydrophilic molecule, it can not pass through cell membrane.
Therefore, the bacterial cells should be capable of uptaking DNA.
This is accomplished by treating them with specific concentration of a divalent cation, e.g., Ca2+ thus making them competent which causes efficient entry of DNA into bacterium through pores in its cell wall.
Recombinant DNA can be forced into such cells by incubating the cells with recombinant DNA on ice, followed by placing them briefly at 42°C (heat shock) and then putting them back on ice. As a result, bacteria gets enabled to pick up recombinant DNA.
There are other methods to introduce foreign DNA into host cells. These are briefly described below.
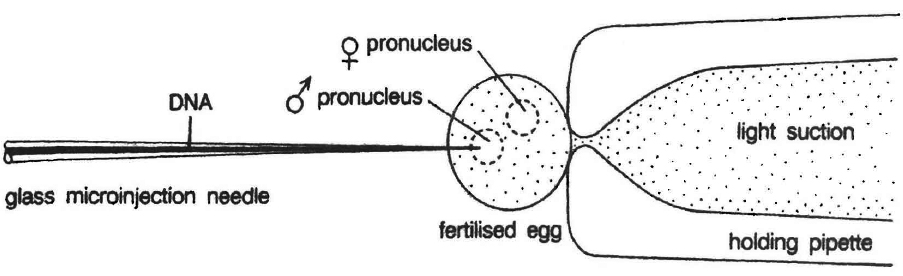
Microinjection
In this method recombinant DNA is directly injected into the nucleus of animal cell by using micro needles or micro pipettes. It is used in oocytes, eggs and embryo. Jeffey S. Chamberlain et. al. (1993) of Human Genome Centre, Michigan University, USA have cured mice that inherited a neuromuscular disease which is like muscular dystrophy of humans.
Direct DNA Injection
Direct injection of DNA into skeletal muscle led to the possibility of using gene as vaccines. Due to low level of expression, therapeutic benefits for the treatment of genetic disorder could not be derived. This method gave birth to the concept of DNA vaccine or genetic immunization.
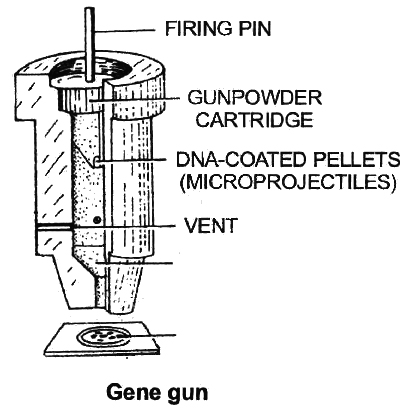
Gene Gun or Biolistics
New technologies like gene gun are also available for vectorless direct gene transfer. DNA coated onto microscopic pellets is literally shot into target cells. Although, it is developed for plants but is also used for animal cells for promoting tissue repair or reducing healing time. This method made great impact in the field of vaccine development.
processes of recombinant DNA technologies
- Books Name
- A TEXT OF BIOLOGY - CLASS XII
- Publication
- ACME SMART PUBLICATION
- Course
- CBSE Class 12
- Subject
- Biology
PROCESS OF RECOMBINANT DNA TECHNOLOGY
Recombinant technology is a complicated process. Several steps lead to the desired goal. The major steps are:
(1) Isolation of DNA,
(2) Digestion of DNA by restriction endonuclease enzyme,
(3) Gene amplification,
(4) Introduction of recombinant DNA into host cells,
(5) Identification of recombinants,
(6) Gene product manufacture, and
(7) Processing.
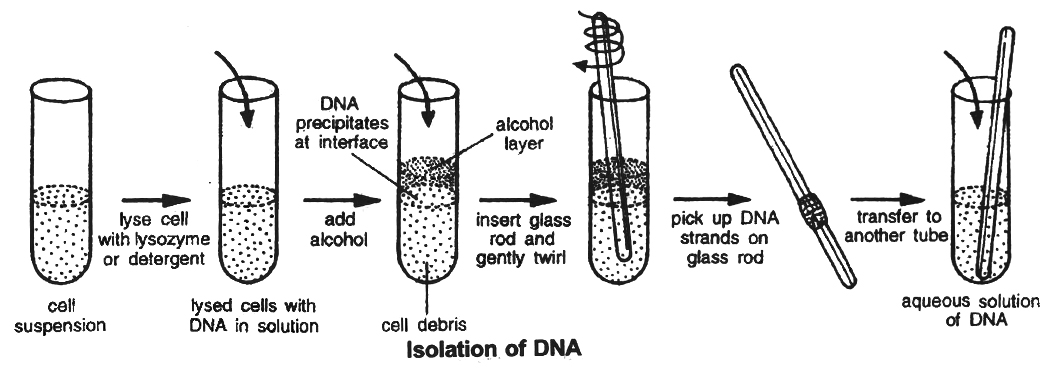
(1) Isolation of DNA: Isolation of the Genetic Material (DNA)
Nucleic acid (DNA or RNA) is the genetic material of all organisms. It is DNA in majority of organisms.
For cutting the DNA with restriction enzymes it needs to be pure and free from other macromolecules.
Because the DNA is covered by the membranes, it has to break the cell open to release DNA and other macromolecules like RNA, proteins, polysaccharides and lipids.
It is obtained by treating the bacterial cells/plant or animal tissue with enzymes such as lysozyme (bacteria), cellulase (plant cells), chitinase (fungus).
As we know that genes are present on long molecules of DNA intertwined with proteins like histones, the RNA can be removed by treating with ribonuclease while proteins can be removed by treating with protease.
Other molecules are removed by proper treatments. The purified DNA finally precipitates out after the addition of chilled ethanol.
This is seen as collection of fine threads in suspension.
(2) DNA digestion by restriction enzymes
The vector and the target DNA fragment can be separately digested with the same restriction enzyme.
The digested vector and the target DNA fragment are then incubated together in the presence of DNA ligase enzyme.
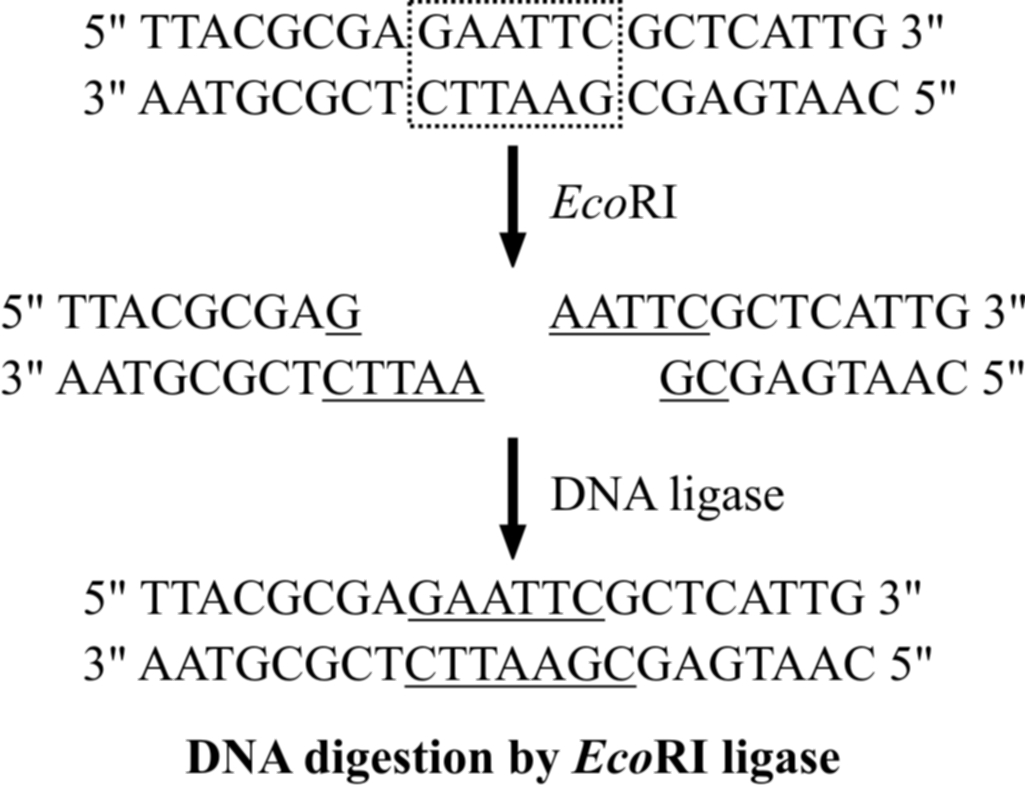
Incubation results in bonding two types of DNA by phosphodiester bonds between them.
Thus, deoxyribose-phosphate backbones of vector molecule and the target DNA fragment are covalently linked, forming a recombinant DNA molecule.
Another possibility in this experiment is the rejoining of the sticky ends of the vector molecule itself, forming a circular vector DNA molecule that is without foreign DNA molecule.
This possibility is eliminated by treating digested, vector with alkaline phosphatase or by using different restriction enzymes.
(3) Gene amplification
This is the process of selective multiplication of a specific region of DNA molecule.
The process has also been used to produce DNA fragments for cloning.
Amplification is achieved by a special method known as polymerase chain reaction (PCR) developed by Kary Mullis in 1985 for which he shared 1993 Nobel Prize.
The principle underlying the technique is to heat double stranded DNA molecule to a high temperature so that the two DNA strands separate into single stranded DNA molecules.
If these single-stranded molecules are copied by a DNA polymerase, it would lead to duplication of the original DNA molecule and if these events are repeated many times, multiple copies of the original DNA sequence can be generated.
The basic requirements of a PCR reaction are the following:
(i) DNA Template: Any source that contains one or more target DNA molecules to be amplified can be taken as template.
(ii) Primers : Primers, which are oligo-nucleotides, usually 10-18 nucleotides long, that hybridize to the target DNA region, one to each strand of the double helix. Two primers are required and these primers are oriented with their ends facing each other, allowing synthesis of the DNA towards one another.
(iii) Enzyme: DNA polymerase which is stable at high temperatures (>90º) is required to carry out the synthesis of new DNA. The polymerase which is generally used in PCR reactions is known as Taq polymerase (isolated from a bacterium Thermus aquaticus). Other thermostable polymerases can also be used.
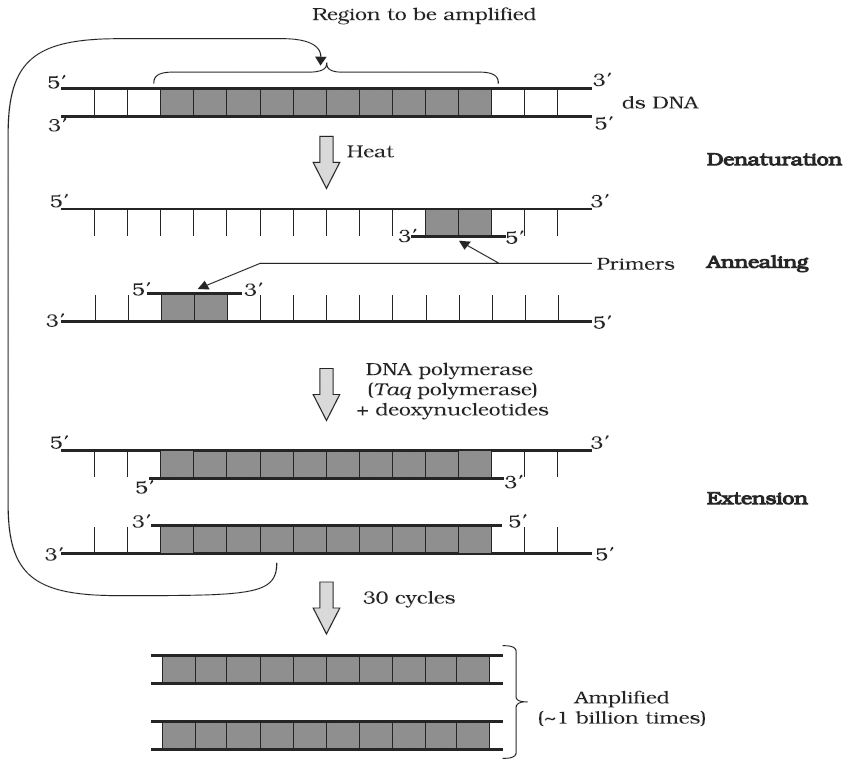
A schematic representation of the three steps performed during PCR. Note that the two primers used are complementary to the 3' end sequences of DNA segment to be amplified
Working Mechanism of PCR :
A single PCR amplification cycle involves three basic steps denaturation, annealing and extension (Polymerisation).
(a) Denaturation: In the denaturation step, the target DNA is heated to a high temperature (usually 94°C), resulting in the separation of the two strands. Each single strand of the target DNA then acts as a template for DNA synthesis.
(b) Annealing: In this step, the two oligo-nucleotide primers anneal (hybridize) to each of the single stranded template DNA since the sequence of the primers is complementary to the 3' ends of the template DNA. This step is carried out at a lower temperature depending on the length and sequence of the primers.
(c) Primer Extension (Polymerisation) : The final step is extension, wherein Taq DNA polymerase (of a thermophilic bacterium Thermus acquaticus) synthesizes the DNA region between the primers, using dNTPs (deoxynucleoside triphosphates) and Mg2+. It means the primers are extended towards each other so that the DNA segment lying between the two primers is copied. The optimum temperature for this polymerization step is 72°C.
To begin the second cycle, the DNA is again heated to convert all the newly synthesized DNA into single strands, each of which can now serve as a template for synthesis of more new DNA. Thus the extension product of one cycle can serve as a template for subsequent cycles and each cycle essentially doubles the amount of DNA from the previous cycle. As a result, from a single template molecule, it is possible to generate 2n molecules after n number of cycles.
Concept Builder
PCR permits early diagnosis of malignant diseases such as leukemia and lymphomas, which is currently the highest developed in cancer research and is already being used routinely.
PCR assays can be performed directly on genomic DNA samples to detect translocation specific malignant cells at a sensitivity which is at least 10,000 fold higher than other methods.
PCR also permits identification of non-cultivatable or slow-growing microorganisms such as mycobacteria, anaerobic bacteria, or viruses from tissue culture assays and animal models.
The basis of PCR diagnostic applications in microbiology is the detection of infectious agents and the discrimination of non-pathogenic from pathogenic strains by virtue of specific genes.
Viral DNA can likewise be detected by PCR.
The primers used need to be specific to the targeted sequences in the DNA of a virus, and the PCR can be used for diagnostic analyses or DNA sequencing of the viral genome.
The high sensitivity of PCR permits virus detection soon after infection and even before the onset of disease.
Such early detection may give physicians a significant lead in treatment.
The amount of virus ("viral load") in a patient can also be quantified by PCR-based DNA quantitation techniques.
Application of PCR
Some of the areas of application of PCR are briefly mentioned here.
(i) Diagnosis of Pathogens : Pathologists use techniques based on detecting specific enzymes or antibodies against disease-related proteins. But these techniques cannot be used for detecting infectious agents that are difficult to culture or that persist at very low levels in infected cells. To overcome these problems, PCR-based assays have been developed that detect the presence of gene sequences of the infectious agents.
(ii) Diagnosis of specific Mutation : PCR can be used to detect the presence of a specific mutation that is responsible for causing a particular genetic disease before the actual onset of the disease. By using PCR, phenylketonuria, muscular dystrophy, sickle cell anaemia, AIDS, hepatitis, chlamydia and tuberculosis can be diagnosed.
(iii) DNA Finger printing: PCR is of immense value in generating abundant amount of DNA for analysis in the DNA fingerprinting technique used in forensic science to link a suspect's DNA to the DNA recovered at a crime scene.
(iv) Detection of Specific Microorganisms: PCR is also used for detecting specific microorganisms from the environment samples of soil, sediments and water.
(v) In Prenatal Diagnosis: It is useful to detect genetic disease in foetus before birth. If the disease is not curable, abortion is recommended.
(vi) Diagnosis of Plant Pathogens : Many diseases of plants can be detected by using PCR. For examples, viroids (associated with apple, grape, citrus, pear, etc.), viruses (like TMV, bean yellow mosaic virus etc.), bacteria, mycoplasmas, etc.
(vii) In Palaeontology: PCR is used to clone the DNA fragments from the mummified remains of humans and extinct animals like wooly mammoth and dinosaurs.
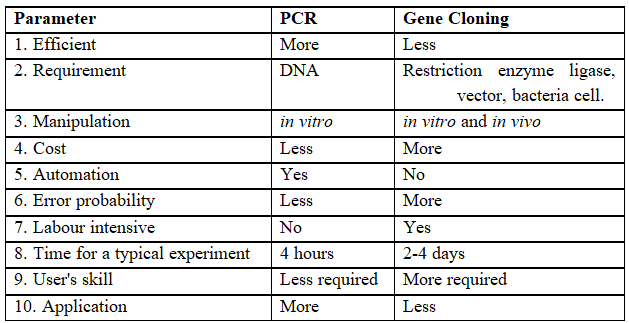
Table-II : Comparison between PCR and Gene Cloning

Concept Builder
Definition of Ligase chain reaction (LCR)
A method of DNA amplification similar to PCR.
LCR differs from PCR because it amplifies the probs molecule rather than producing amplicon through polymerization of nucleotides.
Two probes are used per each DNA strand and are ligated together to 'form' a single probe.
LCR uses both a DNA polymerase enzyme and a DNA ligase enzyme to drive the reaction.
Like PCR, LCR requires a thermal cycler to drive the reaction and each cycle results in a doubling of the target nucleic acid molecule. LCR can have greater specificity than PCR.
Polymerase Chain Reaction (PCR) : Technique for obtaining multiple copies of DNA
(4) Introduction of recombinant DNA into host cells:
Once the recombinant DNA molecule has been multiplied, it needs to be inserted into host cells.
Many methods for introduction are available.
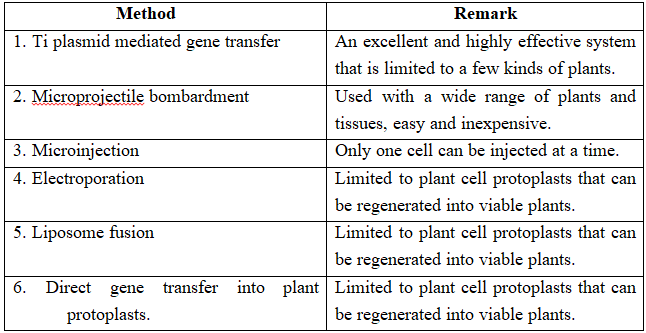
Selection of a method depends upon type of vector and the host cell along with other things.
Some common methods are outlined below-
(a) Transformation: This is a method where cells take up DNA from their surroundings. Since, many cells such as those of E.coli, yeast, mammalian cells, etc. do not naturally absorb DNA, hence they need to be made competent. Mandel and Higa (1970) observed that E. coli cells can be made competent to take up external DNA by suspending them in cold calcium chloride.
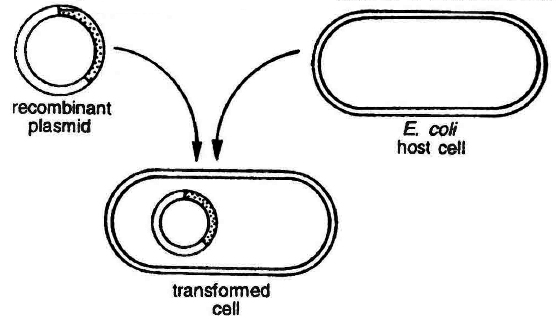
Transformation in bacterial cell
(b) Transfection: In this method DNA is mixed with charged substances like calcium phosphate, cationic liposomes etc. These are spread on the recipient host cells. Calcium ions carry foreign DNA and release it inside the cell since calcium gets precipitated in the form of calcium phosphate, thus transferring the DNA by endocytosis.
(c) Microinjection and macroinjection: Specially designed micromanipulator is used to inject DNA into cytoplasm or the nucleus of a recipient cell or protoplast. The method is used for direct introduction of DNA into plant or animal cells without using special eukaryotic vectors.
(d) Microprojection (biolistics or particle gun) : Tungsten or gold particles (microparticles) coated with DNA are accelerated to a very high initial velocity. These microprojectiles are carried by other (nylon) microprojectiles or the bullet, causing them to penetrate the cell walls of intact target cells or tissues.
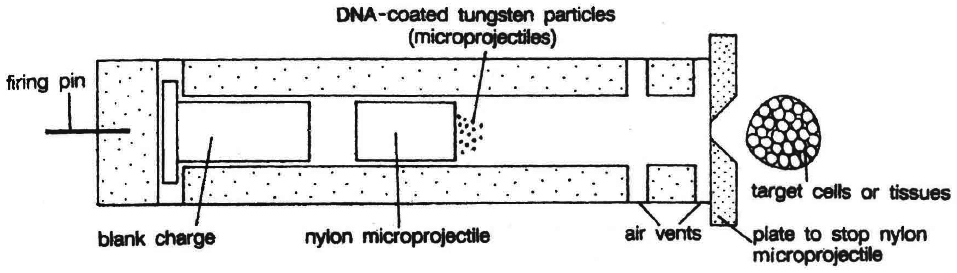
(e) Electroporation: Short electrical impulses of high field strength are given. These increase the permeability of protoplast membrane by creating transient microscopic pores, thus making the entry of DNA molecules into the cells much easier.
(f) Ti plasmid based gene transfer: A more common method of introducing foreign DNA into plant cells is to use the bacterium Agrobacterium tumefaciens and its Ti plasmid.
This gram negative soil bacterium is a plant pathogen and produces crown gall disease in many dicotyledons including grapes, stone fruits, roses, tomato, sunflower, cotton, soyabean, etc.
Most strains of this bacterium carry tumour inducing (Ti) plasmid.
In nature, Agrobacterium attaches to the leaves of the plants and Ti plasmid is transferred into plant cells.
The plasmid becomes incorporated into plant chromosomal DNA.
Therefore, Ti plasmid has been used as a vehicle for introduction of recombinant DNA into plant cells.
Ti plasmids cause tumours in plants.
Strains of the bacterium have been developed which do not have tumour inducing genes.
However, T region of plasmid plays an important role in gene transfer.
This specific segment of bacterial plasmid DNA is called T-DNA (transferred DNA).
T-DNA has a cloning site into which foreign DNA (DNA insert) is inserted.
This recombinant plasmid is now introduced into the bacterium Agrobacterium tumefaciens.
It is then used to infect cultured cells.
The T region of the plasmid with foreign DNA (or DNA insert) is transferred to plant cells.
It gets integrated with chromosomal DNA of the cell.
Cultured cells are induced to grow into plantlets.
These are planted into the soil where the mature plants are formed.
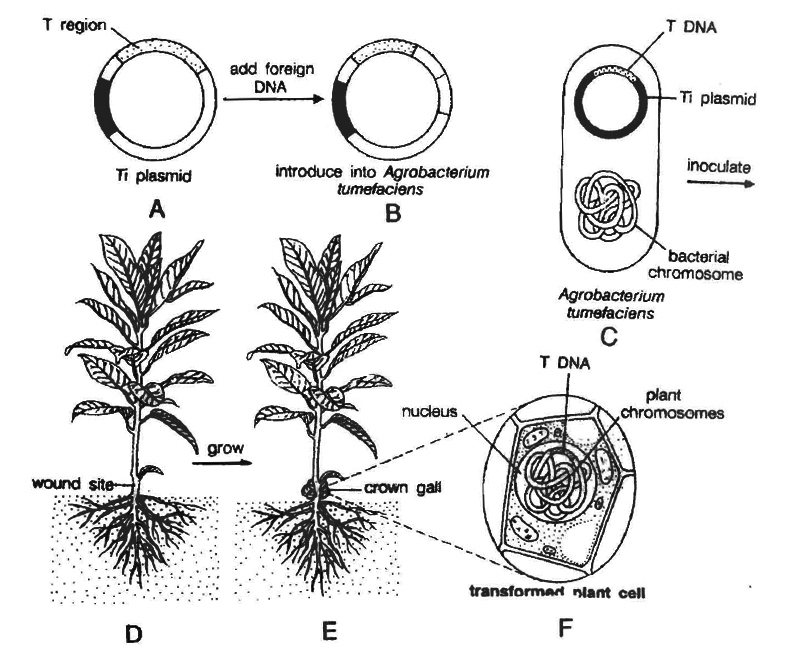
(5) Identification of recombinant :
After the insertion of recombinant DNA into the host cell, these need to be identified from those which do not possess it.
The methods used to do so consider expression or non-expression of certain characters especially antibiotic resistance-gene (e.g., ampicillin resistance gene) on plasmid vector.
Selectable marker usually provides resistance against a substrate which when added to the culture medium, inhibits the growth of normal cells or tissues in culture, so that only transformed tissues will grow.
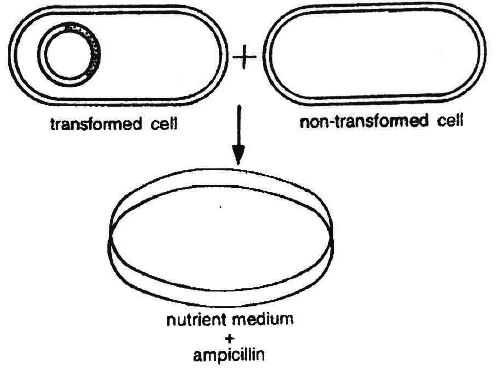
Thus the simplest method for identification is to grow transformed host cells (with ampicillin resistance gene) on medium containing ampicillin.
This would enable the cells containing this transformed plasmid to grow and form colonies.
There are other methods for detection of recombinants based on the fact that the cloned DNA fragment disturbs the coding sequence of gene.
This is known as insertional inactivation.
Let us consider a plasmid containing genes resistant for two different antibiotics, i.e., ampicillin and tetracycline.
If the target DNA fragment is inserted in a site located in ampicillin resistance gene, this gene will then be inactivated.
Thus, host cells with such a recombinant plasmid will be sensitive to ampicillin but resistant to tetracycline.
These host cells will die when grown on ampicillin containing medium but would grow on medium containing tetracycline.
Self ligated or religated (non-recombinant) vectors would grow on medium containing both ampicillin and tetracycline being resistant to them.
Another, but similar method involves insertional inactivation of lac Z gene.
It is known as blue-white selection, being colour based.
(6) Gene product manufacture:
When recombinant DNA is transferred into a bacterial, plant or animal cell, the foreign DNA is multiplied.
Most of the recombinant technologies are aimed to produce a desirable protein.
So there is a need for expression recombinant DNA.
After the cloning of the gene of interest one has to maintain the optimum conditions to induce the expression of the target protein and consider producing it on a large scale.
If any protein encoding gene is expressed in a heterologous host it is known as a "recombinant protein".
The cells having cloned genes of interest can be grown on a small scale in the laboratory.
The cultures may be used for extracting and purifying the desired protein.
The cells can also be multiplied in a continuous system where the used medium is passed out from one side and fresh medium is added from the other side to maintain the cells in their physiologically most active lag exponential phase (Lag phase -no significant increase of the cells, exponential phase -rapid multiplication of the cells).
This type of culturing method produces a larger biomass to get higher yields of desired protein.
Small volume cultures cannot give large quantities of the products.
To produce large quantities of these products, development of "bioreactors" was required where large volumes (100-1000 litres) of culture can be processed, Hence, bioreactors are like vessels in which raw materials are biologically converted into specific products, individual enzymes using microbial, plant, animal or human cells.
A bioreactor provides the optimal conditions for obtaining the desired product by providing optimum growth conditions such as substrate, temperature, pH, vitamins, oxygen and salts.
One of the most commonly used bioreactor is of stirring type.
The presence of stirrer makes mixing possible and also makes oxygen available through the reactor.
A bioreactor also has an agitatory system, an oxygen delivery system, a foam control system, a temperature control system, pH control system and sampling ports so that small volumes of the culture can be withdrawn periodically.
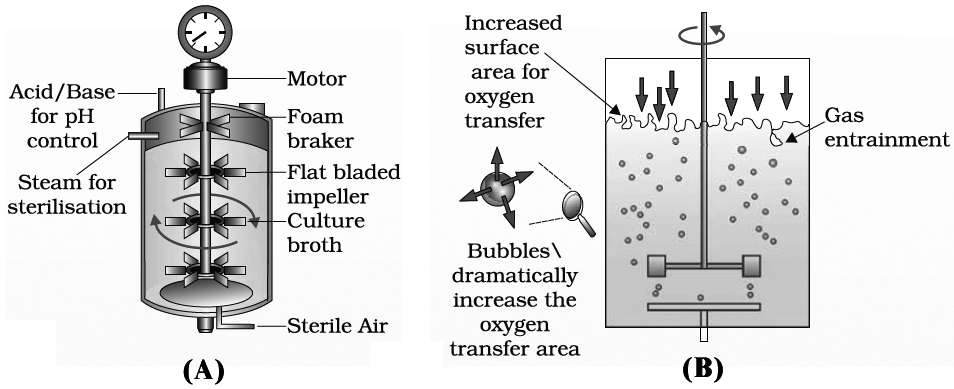
which sterile (free from any germs) air bubbles are sparged.
(7) Downstream Processing :
Once the product is ready, it has to be processed for commercial use.
This requires purification and strict quality control to maintain the efficacy.
The products based on biotechnology must ensure that they satisfy the consumer needs and are not harmful.
Therefore, a thorough checking of products at each level of manufacture is done.
The manufacturing process and the quality control methods vary with each product.
